
Al n团簇(n=1~9)与O2的吸附机理的DFT研究
2011-10-16 11:34崔乘幸唐明生
河南科技学院学报(自然科学版) 2011年2期
崔乘幸,唐明生
(1.郑州大学化学系计算化学研究中心,河南郑州450052;2.河南科技学院,河南新乡453003)
原子和分子团簇,简称团簇(Cluster)或微团簇(Micro-cluster),通常指由几个、几十个乃至上千个原子、分子或者离子通过物理的或化学的结合力组成的相对稳定的非刚性聚集体,其物理和化学性质随所包含的原子数目而变化.在团簇特殊的空间尺寸范围(几十nm到几千nm)内,团簇的许多性质既不同于单个的原子或分子,又不同于液体或固体,而且也不能从单体和体相材料的性质作内插或外延得到.因此,团簇被认为是介于微观原子、分子与宏观凝聚态物质之间物质结构的新层次,称为物质的第5态.1984年,Knight W D对钠团簇的电子层结构和丰度就进行了深入的研究[1].
表面吸附是指在气/固或者气/液两相系统中,分子或者原子在两相界面上的堆积.对于气/固系统,产生吸附的原因在于当气体与固体接触,气体分子(原子)会不断地撞击固体表面,其中有的分子(原子)立即弹回气相,有的则会在表面滞留一段时间,吸附就是这种滞留的结果.
20世纪60年代,Hohenberg、Kohn和sham提出了密度泛函理论[2-3],从而将多电子问题简化为单电子问题,也说明了单电子近似近代理论是在密度泛函理论的基础上发展起来的,同时为分子和固体电子结构总能量的计算提供了有利的工具.近来有很多学者利用密度泛函理论对团簇和小分子之间的吸附作用进行了研究[4-7],本文在前人工作的基础上,运用密度泛函理论方法,采用簇模型对Aln金属簇基态几何结构优化,并对Aln簇(n=1~9)与氧之间相互作用的几何结构、电子结构、吸附能等进行了系统的理论计算研究.
1 实验部分
1.1 实验方案
本文利用Becke型3参数密度泛函模型,此模型采用Lee-Yang-Parr泛函,在6-31++g(d,p)基组条件下对其结构进行优化,确定优化结果中的振动频率全部为正,然后在相同计算方法和基组条件下计算得到吸附能和吸附键长.本实验的全部构型优化,能量计算,频率分析等计算过程全部在Gaussian 03程序包中完成.
1.2 实验结果
1.2.1 Aln团簇的几何结构与键长 Aln团簇在B3LYP/6-31g++g(d,p)条件下进行优化,确定无负频率,即所得构型处于势能面的最低点(Local minimum),然后使用Chemcraft 1.6分子构型软件记录优化结果并标记出团簇的中相应化学键的键长.图1中列出了前5个团簇Aln(n=1~5)优化结果的键长及其稳定构型.

1.2.2 Aln与O2的吸附在Aln(n=1~9)团簇的最稳结构的基础上,我们构建了AlnO2(n=1~9)团簇的桥式结构[9],在相同方法基组条件下对其优化,得到其最稳结构,图2列出了前5个团簇与O2吸附的最稳构型.

图2 Al n O2(n=1~5)优化得到的构型
2 讨论
2.1 Al n O2(n=1~9)团簇的结构与稳定性
为了了解铝团簇与O2吸附结构的稳定性,我们分别计算出了该吸附结构的吸附能和吸附键长,定义吸附能为

其中,EAlnO2,EAln,EO2分别为最稳定结构中AlnO2、Aln、O2的总能量[8].

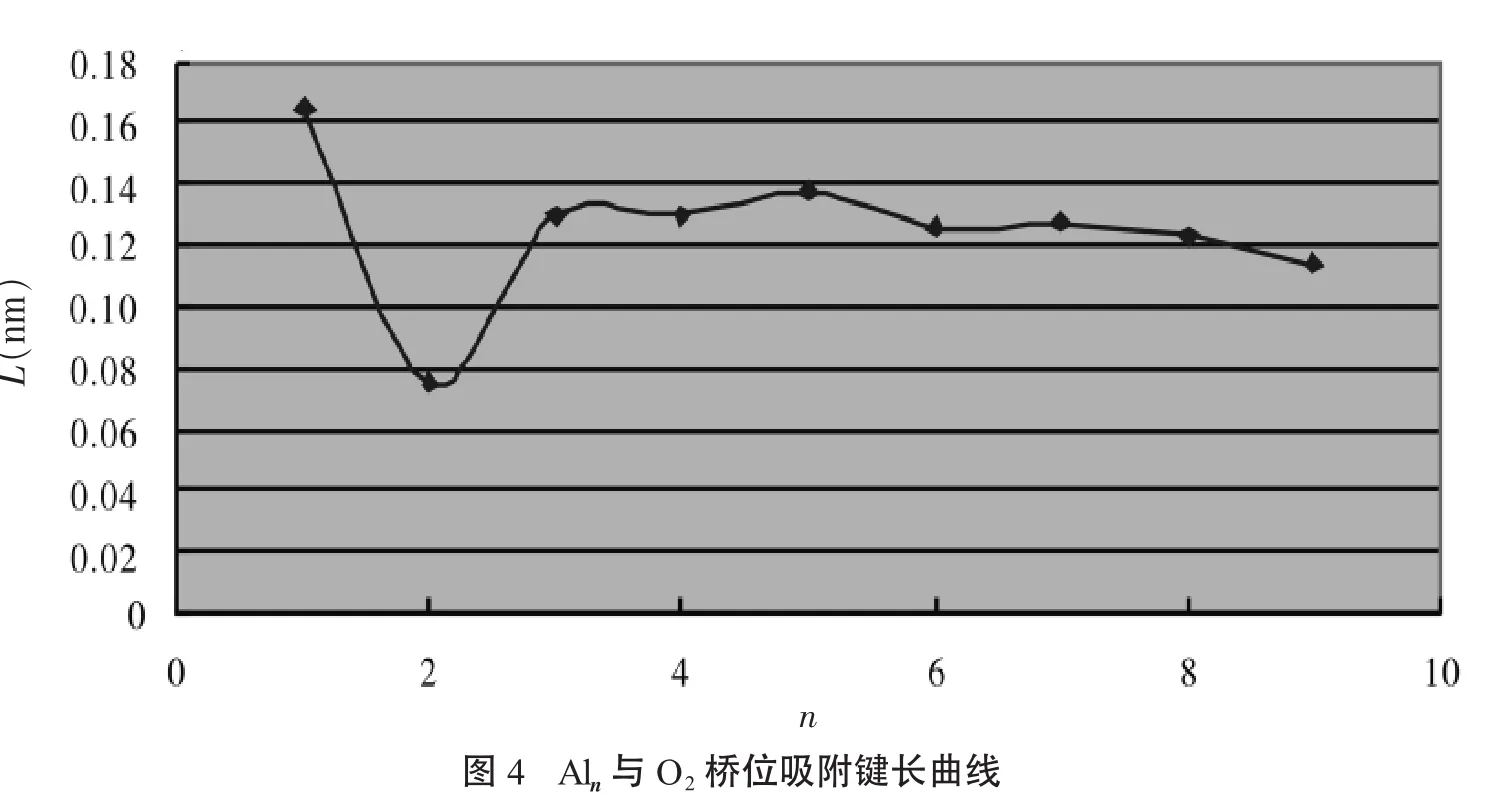
桥位吸附键长L为离吸附键最近的氧原子与与吸附键中点的距离,为了表示方便,我们在该位置添加了一个虚拟原子X,图3、图4给出了Aln与O2的吸附能曲线和Aln与O2桥位吸附键长曲线,通过该曲线可以明显观察到,吸附能都小于-0.05 a.u.,这说明Aln(n=1~9)团簇都能很好的与O2吸附,而Al2的最低,为-0.205 3 a.u.,随后吸附能逐渐升高,但变化不大,都在-0.1 a.u.左右波动,而吸附键长变化也是如此,Al2的吸附键长最短,为0.075 2 nm,随后都比较稳定,在0.13 nm左右,说明Al2O2团簇吸附的最稳定结构相比其他基态团簇而言具有更强的相对稳定性.
2.2 能隙HOMO-LUMO
表1给出了最高占据分子轨道(HOMO)、最低未占据分子轨道(LUMO),以及相应的能级差.HOMO和LUMO的能级之差就是能隙.能隙的大小反映了电子从HOMO向LUMO发生跃迁的能力,在一定程度上体现了团簇分子参与化学反应的能力[9].由表1中亦可以看出,与其他尺寸团簇相比,Al2O2团簇的能隙最大,表明化学活性最低,这也说明Al2O2团簇的化学稳定性较强.

表1 Al n O2(n=1~9)团簇的HOMO、LUMO和能隙
2.3 解离能与平均原子结合能
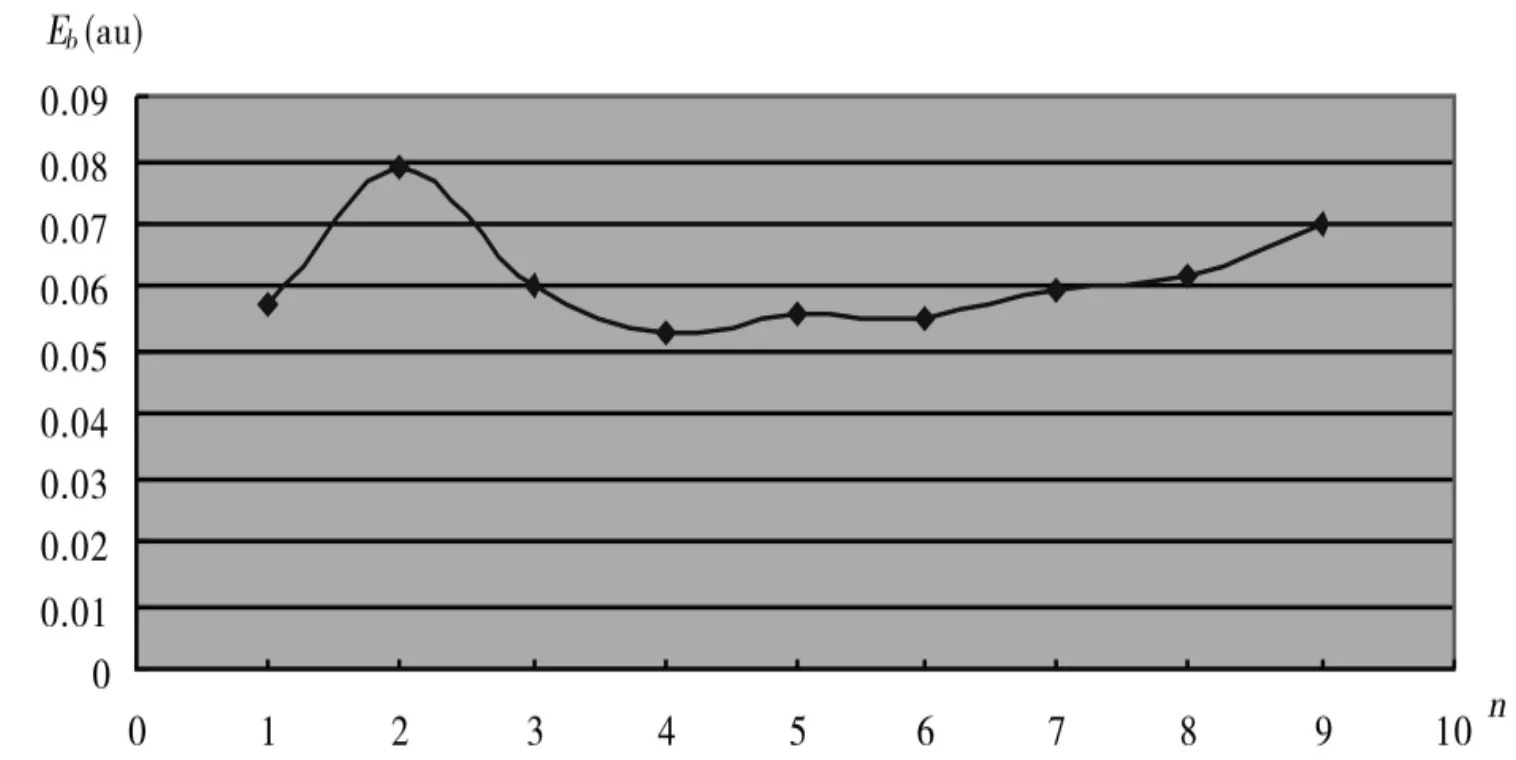
图5 Al n与O2的平均原子结合能
为了清楚地看出引进氧分子对铝团簇的影响,了解最稳定AlnO2(n=1~9)团簇结构的相对稳定性,在考虑自旋多重度的情况下,我们分别计算了AlnO2(n=1~9)的平均原子结合能和解离能.平均原子结合能定义为团簇的结合能除以组成团簇的原子总数;平均原子结合能和解离能的计算公式为
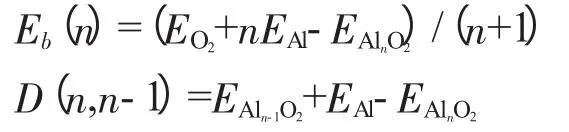
其中EAln-1O2,EAl,EO2分别为最稳定结构中AlnO2、Aln、O2的总能量[10].
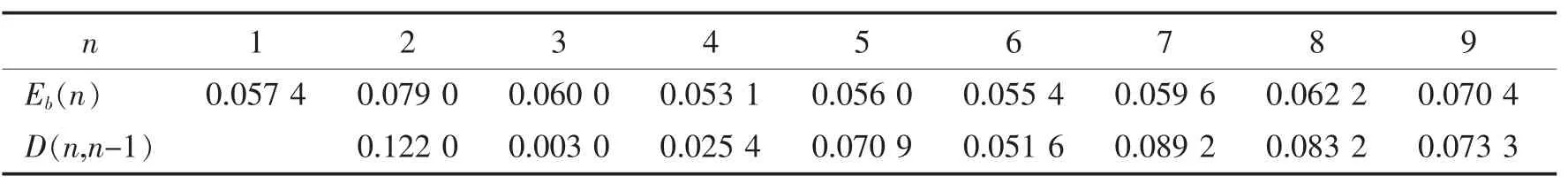
表2 Al n O2(n=1~9)团簇的解离能和平均原子结合能
表2列出了团簇的平均原子结合能和解离能的具体值,D(n,n~1)代表解离能,E(n)表示平均原子结合能.图5和图6分别给出了AlnO2(n=1~9)团簇最稳定结构的平均原子结合能E(n)和解离能D(n,n~1)随团簇尺寸的变化规律.从图5和图6中可以很明显看到,平均原子结合能在n=2时最大.解离能随团簇尺寸的变化趋势和平均原子结合能雷同,也是在n=2时最大.从图5和图6中,不难发现与不同尺寸的最稳定结构相比Al2O2具有最大的解离能和平均原子结合能,这说明Al2O2的最稳定结构相比其他基态团簇而言具有更强的相对稳定性.
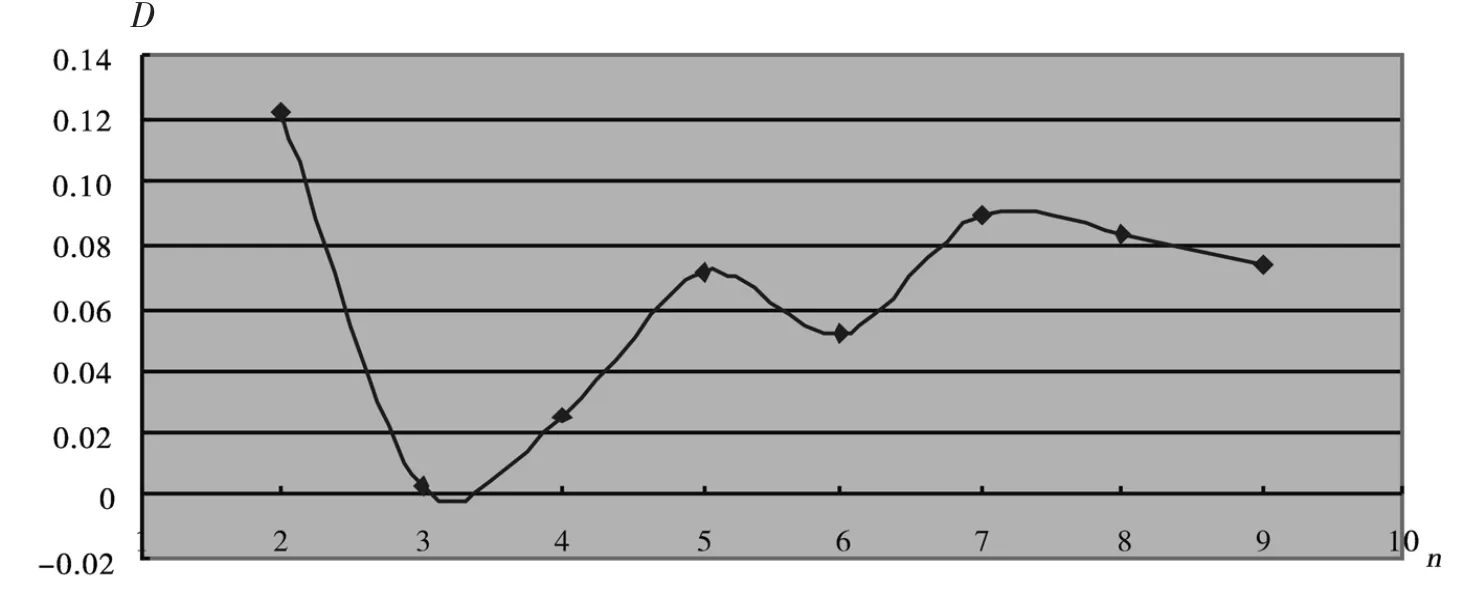
图6 Al n与O2的解离能曲线
3 结论
本文主要在B3LYP/6-31++g(d,p)下,对AlnO2(n=l~9)团簇的吸附能、几何构型以及HOMO-LUMO能隙、解离能与平均原子结合能进行了系统的理论研究.通过上述分析得出以下结论:
(1)AlnO2(n=l~9)团簇都能很好的与O2吸附.
(2)Al2O2不论是在吸附能,桥位吸附键长,还是在HOMO-LUMO能隙,以及解离能与平均原子结合能方面,其最稳定结构与其他基态团簇相比均表现出更强的相对稳定性,故通过对Aln团簇(n=l~9)与O2的吸附机理的研究,我们得出Al2O2团簇具有最大的相对稳定性.
[1]Knight W D,Clemenger K,de Heer W A,et al.Electronic shell structure and abundances of sodium clusters[J].Phys.Rev.Lett,1984,23:2141-2143.
[2]Hohenberg P,Kohn W.Inhomogeneous electron gas[J].Phy.Rev.,1964,136:B864-B871.
[3]Kohn W,Sham L J.Self-consistant equations including exchange and correlation effects[J].Phy.Rev.A,1965,140:1133-1138.
[4]毛丽萍.金团簇与CO、O2和H2O相互作用的第一性原理研究[D].大连:大连理工大学,2009.
[5]曹彪炳,段海明.AnC2(A=Fe,Co,Ni,Cu)团簇的密度泛函研究[J].新疆大学学报,2008,25(4):395-402.
[6]陈颖健.密度泛函理论和量子化学计算[J].国外科技动态,1999(1):11-14.
[7]李责发,彭平,仇治勤,等.Aln[n=2~24,55]团簇结构特性的第一原理计算[J].中国有色金属学报,2006,16(5):823-828.
[8]涂学炎,王登武,陈秀敏.Run(n=2~7)金属团簇与氧反应的DFT研究[J].贵金属,2004,25(3):15-21.
[9]刘丽,刘付轶,韩聚广,等.硼氧小分子在铂团簇表面吸附的密度泛函研究[J].中国科学院研究生院学报,2009,26(2):194-202.
[10]姜勇,储伟,江成发,等.Pdn(n=l~7)团簇及其与甲烷相互作用的密度泛函理论研究[J].物理化学学报,2007,23(11):1723-1727.
猜你喜欢
大学物理(2022年9期)2022-09-28
物理学报(2021年12期)2021-07-01
数学物理学报(2020年6期)2021-01-14
物理通报(2020年7期)2020-07-01
青岛大学学报(工程技术版)(2019年2期)2019-09-10
科技资讯(2016年5期)2016-08-13
武汉工程大学学报(2016年1期)2016-04-07
枣庄学院学报(2015年5期)2016-01-09
中学化学(2015年8期)2015-12-29
原子与分子物理学报(2015年3期)2015-11-24
