
X F2(X=B,N)分子基态的结构与势能函数*
2011-11-02 03:25肖夏杰韩晓琴刘玉芳
物理学报 2011年6期
肖夏杰韩晓琴 刘玉芳
X F2(X=B,N)分子基态的结构与势能函数*
肖夏杰1)2)韩晓琴3)刘玉芳1)
1)(河南师范大学物理与信息工程学院,新乡453007)
2)(河南质量工程职业学院,平顶山467000)
3)(商丘师范学院物理与信息工程系,商丘476000)
(2010年8月9日收到;2010年9月10日收到修改稿)
基于Gaussian03计算软件利用QCISD方法,选用不同基组对X F2(X=B,N)分子基态结构进行了几何优化,在此基础上选出最优基组D95(df,pd)和D95+(df,pd)分别对BF2和NF2分子的谐振频率、力常数等进行了计算.推导出X F2(X=B,N)分子基态的多体展式势能函数,同时根据势能函数绘制了X F2(X=B,N)分子基态的3种等值势能图,从不同角度验证了该势能面符合3原子分子几何构型,清晰准确的再现了X F2(X=B,N)分子基态的结构特征.
BF2,NF2,结构,势能函数
PACS:31.50.Bc,33.15.-e,33.15.Fm
1.引言
氟与第Ⅲ主族元素形成的化合物一直是无机与有机化学研究的热点[1].BF3是有机合成和石油化工广泛应用的一种重要酸性催化剂,在很多有机化学反应过程中都有应用[2],同时BF3是制备卤化硼、元素硼、硼烷、硼氢化钠等的主要原料,对BF3理论和实验研究一直较多[3].NF3气体在高能化学激光、电子工业以及太阳能光电产业等方面具有非常广泛的应用,同时NF3具有在等离子条件下释放氟的能力,是一种热力学稳定的氧化剂[4];随着科技的飞速发展,对NF3理论和实验的研究受到人们高度的重视[5,6].研究BF,BF2,NF,NF2分子的结构与性质是得到BF3,NF3分子势能函数与相关动力学参数的基础.1972年Colin等[7]用从头计算的多组态自洽场理论(MCSCF)研究了BF2,NF2的结构和原子化能,1981年Kurtz小组[8]用相对论Hartree Fock(RHF)理论研究了BF的基态分子结构,1993年Papakondylis等[9]研究了NF2的分子结构,1999年Charles等[3]用B3LYP/6-311+G(2df)方法研究了BF,BF2,BF+, BF+2的结构、谐振频率、电离能和原子化能,2003年彭谦等[10]用密度泛函方法计算了BF的电子结构与光谱数据,2005年谢安东、朱正和等[2]利用SAC/ SAC方法,对BF分子的势能函数进行了研究.2008年Jiri Czernek等[11]研究了NF2的分子结构和离解能.尽管还有对BF,BF2,NF,NF2基态结构的实验与理论研究结果[12—22],但均未涉及BF2,NF2力常数和势能函数的研究,用QCISD方法对BF2,NF2分子基态势能函数的研究,未见报道.
本文基于Gaussian03计算软件利用QCISD方法,分别选用D95(df,pd)和D95+(df,pd)基组对BF2,NF2分子基态的平衡几何、谐振频率、力常数等进行了计算,并在此基础上推导出了BF2,NF2分子的多体展式势能函数,同时根据势能函数讨论了势能面的静态特征.
2.理论计算与结果讨论
2.1.X F2(X=B,N)分子基态的平衡结构、振动频率与力常数
基于Gaussian03计算软件利用QCISD方法,选常用基组分别对BF2,NF2分子进行结构优化,得到其键长RF—X、键角α、离解能De见表1.同时得到它们的基态构型都为C2v,电子态分别为X2A1[3,7,21]和X2B1
[7,9,11],与文献一致.

表1 X F2(X=B,N)基态几何优化计算结果
表1中不同基组计算得到的键长RX—F、键角α与实验结果比较,可以看出BF2分子采用D95(df,pd)基组和NF2分子采用D95+(df,pd)基组得到的结果最接近实验值.分别使用这两个基组对BF2,NF2分子基态进一步计算,得到振动频率ν(对称伸缩振动频率ν1(α1)、弯曲振动频率ν2(α1)、反对称伸缩振动频率ν3(b1))和力常数f见表2.

表2 X F2(X=B,N)的平衡结构与谐振频率
2.2.X F2(X=B,N)分子基态的多体项展式势能函数
根据原子分子反应静力学原理[13],推导出BF2(X2A1)、NF2(X2B1)可能的离解极限分别为

对于基态X F2(X=B,N)分子,以R1,R2,R3分别表示X—F,F—X,F—F的间距,选择基态原子能量为零,则满足上述离解极限的多体项展式为
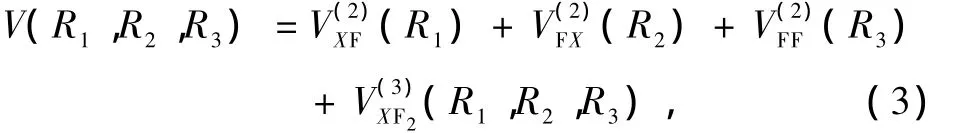
其中R1=R2=RXF,R3=RFF,V(X2F)(R1),V(F2X)(R2), V(F2F)(R3)分别为两体项BF(X1Σ+)或NF(X3Σ-), F2(X1Σ+)的势能函数,V(3)XF2(R1,R2,R3)为三体项BF2(X2A1),NF2(X2B1)的势能函数,两体项与三体项势能函数的计算结果如下:
2.2.1.两体项BF,NF的势能函数
为了得到基态X F2(X=B,N)分子的多体项展式势能函数,本文对双原子分子采用Murrell-Sorbie势能函数形式:
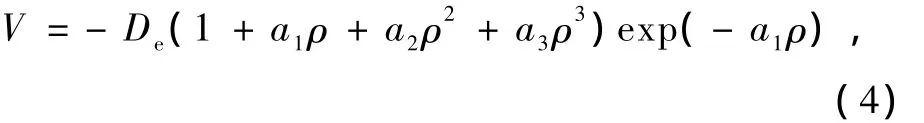
其中De为双原子分子的离解能,ρ=R-Re,而R,Re分别为双原子分子的核间距和平衡核间距,a1,a2,a3为拟合参数.
分别采用QCISD,CCSD,B3 LYP,B3 P86等方法,选常用基组D95+(df,pd),D95+(2 df,pd),6-311++g(df,pd),6-311++g(2 df,pd),6-311 ++g(3df,2 pd),6-311++g(3 df,3 pd)等对BF (X1Σ+),NF(X3Σ-)分子进行几何优化和频率计算.通过与文献值、实验值的对比,发现对BF (X1Σ+)用CCSD/6-311++g(3 df,3 pd)方法计算得到的结果最优,对NF(X3Σ-)用B3 LYP/6-311 ++g(df,pd)方法计算得到的结果最优.在此基础上分别对BF(X1Σ+),NF(X3Σ-)进行单点能扫描,进一步将扫描结果拟合为Murrell-Sorbie势能函数.BF(X1Σ+),NF(X3Σ-),F2(X1Σ+)基态的Murrell-Sorbie势能函数的各项参数见表3.由表3可以看出拟合得到的BF(X1Σ+),NF(X3Σ-)基态的Murrell-Sorbie势能函数的各项参数与实验值相比达到了较高的精度.
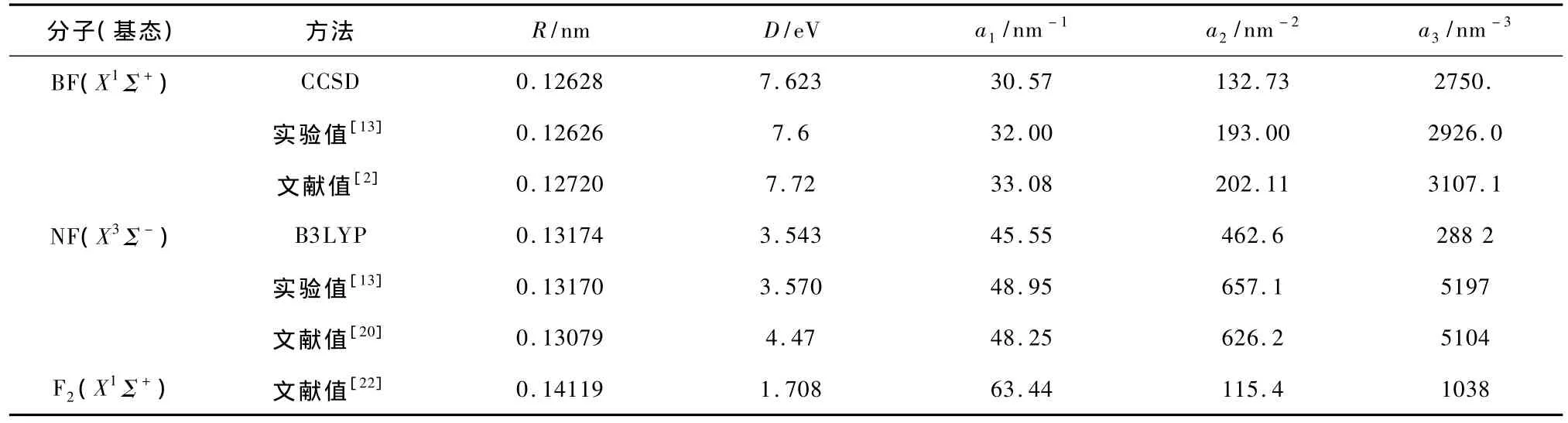
表3 BF(X1Σ+),NF(X3Σ-),F2(X1Σ+)的Murrel-Sorbie势能函数参数
图1和图2分别表示出了BF(X1Σ+),NF(X3Σ-)分子基态的势能曲线.其中,实线为拟合得到的Murrel-Sorbie势能函数曲线,圆圈线为单点能扫描曲线.图1,图2中,拟合函数曲线与单点能扫描曲线比较,相应位置处的点、线之间符合得较好,说明拟合得到的Murrell-Sorbie势能函数能够正确地反映BF(X1Σ+),NF(X3Σ-)分子基态.由Murrell-Sorbie势能函数与力常数的关系以及力常数与光谱数据的关系,可以分别得到BF(X1Σ+),NF(X3Σ-),F2(X1Σ+)的光谱数据,结果见表4.由表4可知,计算得到的BF(X1Σ+),NF(X3Σ-)的光谱数据与实验值相比达到了较高的精度.

图1 BF基态的势能曲线
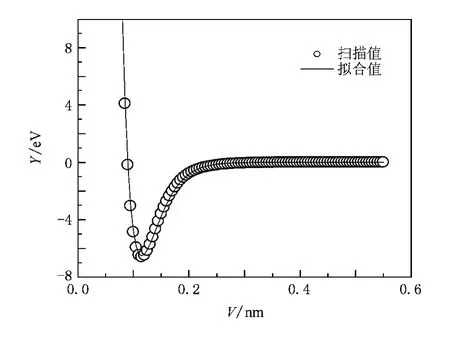
图2 NF基态的势能曲线

表4 BF(X1Σ+),NF(X3Σ-),F2(X1Σ+)的光谱常数
2.2.2.三体项BF2,NF2的势能函数
三体项BF2(X2A1),NF2(X2B1)的势能函数,采用文献[22—25]中的形式:

其中,P为对称内坐标Si的多体项,T为量程函数,它们的形式为
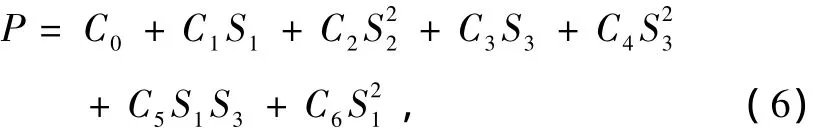

基态BF2(X2A1),NF2(X2B1)的平衡构型都为C2v对称结构,为了方便研究其势能函数,根据势能面的结构特征,采用优化内坐标.取X F2(X=B,N)的两个平衡键长为参考结构,计算中所使用的内坐标ρi经以下变换成为优化内坐标Si:

式中ρi=Ri-R0i(i=1,2,3),其中S2对R1和R2的交换是反对称的,为了满足这一物理意义,S2只能含偶次项.
由以上可知,要得到三体项BF2(X2A1),NF2(X2B1)的势能函数V(3)XFx2(R1,R2,R3)就必需确定7个线性系数(C0,C1,C2,C3,C4,C5,C6)和两个非线性系数λ1,λ2.对势能面进行非线性优化,可以确定出两个非线性系数λ1,λ2,根据表2列出的BF2(X2A1),NF2(X2B1)的平衡结构常数、离解能及力常数可以确定出(6)式中的7个线性系数(C0,C1,C2,C3,C4,C5,C6).结果见表5.
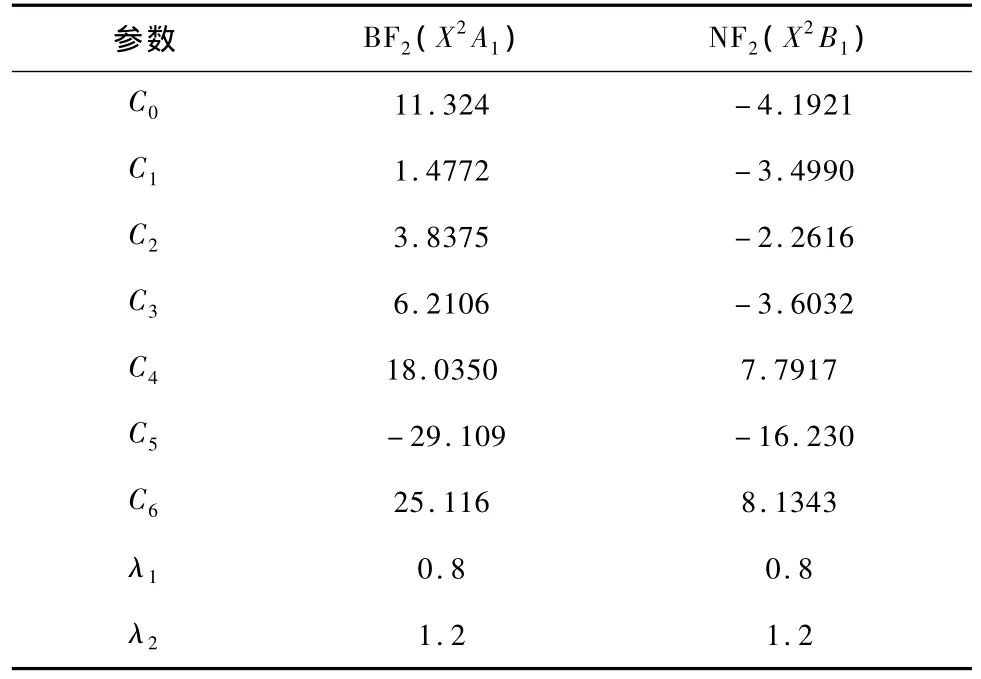
表5 BF2(X2A1),NF2(X2B1)的解析势能函数的三体项参数
根据基态X F2(X=B,N)分子的多体项展式势能函数,进一步可绘制出其等值势能图,如图3—8所示.图像清晰地反映了X F2(X=B,N)分子基态的平衡结构特征.
图3是根据BF2(X2A1)的解析势能函数,在固定∠FBF=122.928°的情况下,得到F—B键对称伸缩振动的等值势能图.图中很好地反映了BF2分子基态的离解能及平衡结构特征.在平衡点RBF=RFB=0.131 nm位置处有一势阱出现,阱深约为3.9374 eV,由此说明在该处形成稳定的BF2分子,这与结构优化结果及得到的离解能计算结果相符.在BF+ F→BF2两个等价的通道上存在两个对称的鞍点,位于(0.14789,0.24263)和(0.24263,0.14789)处,活化能大约为30.875 kJ/mol,并且在RBF=RFB= 0.227 nm位置处有一势垒,势垒高度约为0.7 eV,说明反应只能通过两个等价的通道越过势垒才能进行.

图3 BF2分子基态的伸缩振动势能图(等值线的间隔为0.38 eV)
图4 和图5分别为F原子绕B—F键与B原子绕F—F键旋转时形成的等值势能图,它们都能表明在RBF=0.227 nm,∠FBF=122.928°处,BF2分子的离解能最低,约为3.9374 eV.由此可以说明在该处形成稳定的BF2分子,与结构优化结果及得到的离解能计算结果相符.
图6是根据NF2(X2B1)的解析势能函数,在固定∠FNF=103.35°的情况下,得到F—N键对称伸缩振动的等值势能图,很好地反映了NF2分子基态的离解能及平衡结构特征.由图6可知,NF2分子基态的平衡结构RN—F=RF—N=0.135 nm位置处有一势阱出现,阱深约为4.7351 eV,由此可以说明在该处形成稳定的NF2分子,准确再现了分子基态的C2v结构,这与结构优化结果及得到的离解能计算结果相符,并且在NF+F→NF2反应中不存在鞍点,该反应为无阈能反应.
图7是F原子绕N—F键旋转时形成的等值势能图,即固定N—F键在X轴上,以N—F键的中点为原点建立坐标系.由该图可以看出,当F原子旋转到距离N原子约0.135 nm处且∠FNF=103.35°时,NF2分子的离解能最低,约为4.7351 eV.

图4 F原子绕B—F键旋转时的等值势能图(等值线的间隔为0.5 eV)

图5 B原子绕F—F键旋转时的等值势能图(等值线的间隔为0.4 eV)

图6 NF2分子基态的伸缩振动势能图(等值线的间隔为1 eV)

图7 F原子绕N—F键旋转时的等值势能图(等值线的间隔为1 eV)
图8 是N原子绕F—F键旋转时形成的等值势能图,即固定F—F键在X轴上,以F—F键的中点为原点建立坐标系.该图反映了当N原子旋转到(0.000 nm,0.135 nm)位置时,NF2分子的能量最低,约为4.7351 eV.由此可以说明在该处形成稳定的NF2分子,准确地再现了NF2分子基态的C2v结构,这与结构优化结果及得到的离解能计算结果相符.

图8 N原子绕F—F键旋转时的等值势能图(等值线的间隔为1 eV)
BF2和NF2分子都是C2v结构且键长、键角相差无几,但其形成分子的机理完全不同,BF+F→BF2反应为越过势垒才能进行的有阈能反应,NF+F→NF2为容易进行的无阈能反应.
3.结论
基于Gaussian03计算软件利用QCISD方法,分X F(X=B,N)分子基态的平别选用不同的基组对2衡几何、谐振频率、力常数等进行了计算,进一步计算得到了X F2(X=B,N)分子基态的离解能,并在此基础上推导出了X F2(X=B,N)分子基态的多体展式势能函数,同时根据势能函数绘制了X F2(X=B,N)分子基态的等值势能图进而讨论了势能面的静态特征.X F2(X=B,N)分子基态都为C2v结构,BF2分子的基电子态为X2A1,平衡核间距为0.13138 nm,键角为122.928°,NF2分子的基电子态为X2B1,平衡核间距为0.13532 nm,键角为103.35°.根据势能函数绘制的X F2(X=B,N)分子基态的3种等值势能图从不同角度验证了该势能面符合三原子分子的几何构型,清晰准确地再现了X F2(X=B,N)分子基态的结构特征.同时等值势能图还反映出X F2(X=B,N)形成分子的机理完全不同.这些不但为研究X F+F(X=B,N)体系的分子反应动力学提供了理论依据,同时也为X F3(X=B,N)的理论和实验研究奠定了理论基础.
[1]Yu H T,Huang X R,Chi Y J 2001 Acta Chim.Sin.59 1055 (in Chinese)[于海涛、黄旭日、池玉娟2001化学学报59 1055]
[2]Xie A D,Zhu Z H 2005 Acta Chim.Sin.63 2126(in Chinese)[谢安东、朱正和2005化学学报63 2126]
[3]Charles W,Bauschlicher Jr,Alessandra Ricca 1999 J.Phys.Chem.A 103 313
[4]Peng L P,Wang S B 2007 Chem.Ind.Engng.24 86(in Chinese))[彭立培、王少波2007化学工业与工程24 86]
[5]Zhang H B,Wei G H,Zhou S R 2001 J.Chin.Mass Sp.Soc.22 51(in Chinese)[张洪彬、韦桂欢、周升如2001质谱学报22 51]
[6]Du W H 2009 Low Temp.Spec.Gas.27 14(in Chinese)[杜伟华2009低温与特气27 14]
[7]Colin T,Douglas A B 1972 Chem.Phys.Lett.16 573
[8]Kurtz H A,Jordan K D 1981 Chem.Phys.Lett.81 104
[9]Papakondylis A,Mavridis A 1993 Chem.Phys.Lett.216 167
[10]Peng Q,Zhai G H,Wang Y B 2003 Acta Chim.Sin.61 1375 (in Chinese)[彭谦、翟高红、王育彬2003化学学报61 1375]
[11]Jiri Czernek,Oldrich Zivny 2008 Chem.Phys.Lett.457 54
[12]Moeller M B,Stuart J 1973 Chem.Phys.Lett.19 78
[13]Zhu Z H,Yu H G 1997 Molecular Structuer and Potential Energy Function(Bering:Science Press)p102(in Chinese)[朱正和、俞华根1997分子结构与分子势能函数(北京:科学出版社)第102页]
[14]Huber,K P,Herzberg G 1979 Molecular Spectrum and Molecular Structure(Vol.4)(New York:van Nostrand Reinhold Company)250
[15]Yu H T,Huang X R,Chi Y J 2002 Chim J Chin.Univ.23 888 (in Chinese)[于海涛、黄旭日、池玉娟2002高等化学学报23 888]
[16]Muller H S P,Loblein K 2008 J.Mol.Spectrosc.251 185
[17]Karthikeyan B,Bagare S P,Rajamanickam N,Raja V 2009 Astropart.Phys.31 6
[18]Rode M F,Sadlej J 2002 Chem.Phys.Lett.358 237
[19]Brown R D,Burden F R,Godfrey P D 1974 J.Mol.Spectrosc.52 301
[20]Liu Y C,Jiang G,Zhu Z H 2003 Acta Phys.Chim.Sin.19 858 (in Chinese)[刘幼成、蒋刚、朱正和2003物理化学学报19 858]
[21]Yoshimine M,McLean A D M 1967 Int.J.Quantum Chem.1 313
[22]Jiang L J,Liu Y F,Liu Z Z,Hang X Q 2009 Acta Phys.Sin.58 201(in Chinese)[蒋丽娟、刘玉芳、刘振中、韩晓琴2009物理学报58 201]
[23]Shi D H,Zhang J P,Sun J F,Liu Y F,Zhu Z L 2009 Acta Phys.Sin.58 5329(in Chinese)[施德恒、张金平、孙金峰、刘玉芳、朱遵略2009物理学报58 5329]
[24]Zhang J P,Shi D H,Sun J F 2008 J.Atom.Mol.Phys.25 1404(in Chinese)[张金平、施德恒、孙金峰2008原子与分子物理学报25 1404]
[25]Han X Q,Jiang L J,Liu Y F 2010 Acta Phys.Sin.59 1000(in Chinese)[韩晓琴、蒋丽娟、刘玉芳2010物理学报59 1000]
PACS:31.50.Bc,33.15.-e,33.15.Fm
*Project supported by the National Natural Science Foundation of China(Grant No.60977063),the Innovation Scientists and Technicians Troop Construction Projects of Henan Province,China(Grant No.084100510011),and the Innovation Talents Program of Institution of Higher Education of Henan Province,China(Grant No.2006 KYCX002).
Corresponding author.E-mail:yf-liu@henannu.edu.cn
Structure and potential energy function of X F2(X=B,N)molecular ground state*
Xiao Xia-Jie1)2)Han Xiao-Qin3)Liu Yu-Fang1)
1)(Collegel of Physics and Information Engineering,Henan Normal University,Xinxiang 453007 China)
2)(Henan Quality Polytechnics,Pingdingshan 467000 China)
3)(Department of Physics and Information Engineering,Shangqiu Normal College,Shangqiu 476000 China)
(Received 9 August 2010;revised manuscript received 10 September 2010)
Based on the Gaussian03 calculation software,QCISD method is used to optimize the possible ground-state structures of X F2(X=B,N)molecules with the different basis sets.On this basis,the resonant frequencies and force constants are calculated with the D95(df,pd)and D95+(df,pd)basis sets respectively.The potential energy functions of X F2(X= B,N)are derived from the many-body expansion theory.At the same time,according to the potential energy functions,three equivalent potential energy diagrams for the X F2(X=B,N)ground states are drawn.The potential energy surface is verified to be consistent with the three atomic molecular geometry.The structures and the features of X F2(X=B,N)can correctly reappear on the potential surface.
BF2,NF2,structure,potential energy function
*国家自然科学基金(批准号:60977063)、河南省创新型科技人才队伍建设工程(批准号:084100510011)、河南省高等学校杰出科研人才创新工程(批准号:2006 KYCX002)资助的课题.
.E-mail:yf-liu@henannu.edu.cn
猜你喜欢
——《势能》
文化纵横(2022年3期)2022-09-07
中学生数理化·八年级物理人教版(2022年6期)2022-06-05
数学物理学报(2022年3期)2022-05-25
数学物理学报(2022年1期)2022-03-16
中学生数理化·八年级物理人教版(2021年6期)2021-11-22
数学物理学报(2021年5期)2021-11-19
数学物理学报(2021年3期)2021-07-19
防爆电机(2020年5期)2020-12-14
中学生数理化·八年级物理人教版(2019年6期)2019-06-25
电测与仪表(2016年14期)2016-04-11
