
钴磷族化合物Ba T2P2和Ba T2As2(T=Co,Rh,Ir)的电子结构*
2011-11-02 03:25钱玉敏徐刚
物理学报 2011年6期
钱玉敏徐刚
钴磷族化合物Ba T2P2和Ba T2As2(T=Co,Rh,Ir)的电子结构*
钱玉敏徐刚
(北京凝聚态物理国家实验室,中国科学院物理研究所,北京100190)
(2010年7月12日收到;2010年9月9日收到修改稿)
基于密度泛函理论(DFT)在广义梯度(GGA)下计算了钴磷族化合物Ba T2P2和Ba T2As2(T=Co,Rh,Ir)的电子结构.研究发现在BaCo2P2和BaCo2As2中,由于范霍夫畸点位于费米面附近使得费米能级处的态密度非常高,从而导致由斯通纳机理引起的巡游铁磁不稳定性.在从Co到Rh然后到Ir的化合物中由于d轨道的空间扩展性增强,直接的d-d以及d-p杂化都会增强,这使得形成范霍夫畸点的d-d:σ键的反键态被推往远离费米能级之上更高的能量区间,从而使得在费米面处的态密度下降,斯通纳机理消失,从而使得Rh和Ir的化合物都稳定在非磁态.本文仔细研究并比较了这些化合物间的电子结构及其演化,计算表明自旋-轨道耦合相互作用对电子结构影响很小.
电子结构,范霍夫畸点,斯通纳不稳定性
PACS:71.20.-b,71.18.+y,75.20.-g
1.引言
最近Fe-基超导体的发现在学术界引起了巨大的反响[1—7],它提供了一类非Cu元素的高温超导材料,并展示出了许多与Cu基超导体不同的新奇物理现象.其中有关磁性对其电子结构的影响以及它与超导之间关系的研究尤为重要[8—10].Fe基超导体与Cu基超导体电子结构的不同主要来自于两方面:1)Fe基超导体是典型的多轨道系统,而Cu基超导体可以用有效的单带模型描述;2)Fe基超导体中的电子关联效应处于中间层次,不很强也不很弱,而Cu基超导体是典型的强关联电子系统.这些不同都给电子结构的研究提出了更强的挑战.本文将讨论钴磷族化合物的电子结构和磁性问题.
对于铁磷族化合物的晶格、磁性、超导等已有很多研究.LaFePO属于非磁超导体,超导转变温度为7 K[11];LaFeAsO在130 K以下呈反铁磁的条纹相[12,13],局域磁矩为0.36μB/Fe,比起处于高自旋状态下3 d6的饱和磁矩4μB/Fe要小很多,显示出由于电子间相互作用引起的重整化效应很明显; BaFe2As2随着温度降低到140 K,也会发生晶格转变并伴有反铁磁自旋密度波的出现,局域磁矩为1.1μB/Fe[14].相对于铁磷族化合物,镍磷族化合物通常都是顺磁态金属,而且掺杂后的超导转变温度比较低,顺磁体LaNiPO[15,16],LaNiAsO[15,16],BaNi2As[15,16]2和BaNi2P[17—19]2的超导转变温度都不超过5 K,第一性原理计算电声耦合系数表明镍磷族都是常规超导体,这与铁磷族化合物很不一样.另一方面,钴磷族体系处于铁磷族和镍磷族之间,实验上没有观测到超导现象.LaCoPO[20]和LaCoAsO[20]在低温下都是铁磁态,居里温度分别为TC=43,66 K,饱和磁矩为0.33,0.39μB/Co.第一性原理计算能够很好描述其铁磁态,都属于斯通纳不稳定机理引起的巡游铁磁态.对于BaCo2As[21]2,在低温下(到1.8 K)实验上并没有观察到铁磁长程序的出现.但是60 K以下磁化率突然增加的现象以及第一性原理计算得到铁磁态结果都表明,它处于铁磁态边缘.对于BaRh2As[22]2
实验报道了顺磁磁化率各向异性,但一直到2 K都没有出现超导或者磁性相变.对于BaIr2As2和BaCo2P2来说,到目前为止还没有任何实验数据.最近,在BaRh2P2和BaIr2P2实验中发现了超导现象[23],其超导转变温度分别为Tc=1.0,2.1 K,这促使我们去系统的研究钴磷族化合物的电子结构以及其中可能存在的磁性不稳定性.
本文将讨论122结构中从Co到Rh,Ir,体系随d轨道序数(从3d到5 d)变化时电子结构及其磁性的演化规律.着重讨论如下3个问题:1)这些化合物是否都是处于铁磁态边缘?2)铁磁不稳定性和电子结构是如何演化的?3)自旋-轨道耦合将会对体系电子结构产生怎样的影响?
2 .晶体结构的优化和计算细节
这里所有的122化合物都具有四方晶胞ThCr2Si2类型的结构(空间群I4/mmm),其结构如图1所示.整个晶格是由边界相连的TX4(T=Co,Rh,Ir;X=As,P)四面体层(在ab平面)和Ba原子层沿c轴方向相互交叠形成.每一个过渡金属离子T都由四个配位体阴离子X包围形成一个四面体.计算选取晶格基矢作为坐标轴.故在这种晶体场中5度简并的d轨道实际上被分为t2g(x2-y2,yx,xz)和eg(xy,3 z2-1)轨道.由于BaCo2P2和BaIr2As2没有关于其晶格参数和原子内部坐标的实验数据,故假设它们同样具有ThCr2Si2类型的结构,并对其晶格结构进行了优化.

图1 ThCr2Si2类型(a)晶格结构;T表示钴族过渡金属元素T(T=Co,Rh,Ir),X表示磷族元素(X=P,As);(b)布里渊区及其高对称点

表1 Ba T2P2和Ba T2As2优化后的晶格参数和文献[21—24]的实验值
我们通过基于赝势平面波方法的BSTATE[25]软件包对这些钴磷族化合物都进行了计算.计算中采用了超软赝势和广义梯度近似(GGA)的交换关联势[26].波函数的截断能量为350 eV,电荷的截断能为2800 eV,计算中采用修正的四面体积分方法,布里渊区划分格点为12×12×16.我们首先优化了晶格结构.优化后的晶格参数和实验晶格参数都列在表1中,除了BaCo2As2以外,其他化合物优化的晶格参数和实验晶格参数符合得很好.正如在以往密度泛函理论(DFT)对LaFeAsO的晶格优化中As原子的位置和实验有较大出入[27],有观点认为这是由于自旋涨落在DFT计算中被忽略.该偏差说明BaCo2As2中自旋涨落效应也很明显.
同时还计算了Pauli磁化率γ0和比热系数χ0,对于BaT2P2,计算值和实验值能很好地符合,显示BaRh2P2和BaIr2P2的电子结构都是属于常规的费米液体范畴.对于BaCo2As2计算的Pauli磁化率比实验值要小很多,结合实验显示它位于铁磁边缘,自旋涨落剧烈导致计算和实验的偏差.对于BaRh2As2计算的Pauli磁化率和比热系数都比实验值要大,和以往的计算一样[28].
3 .Ba T2P2和Ba T2As2(T=Co,Rh,Ir)的电子结构
在ThCr2Si2结构的化合物中一个很重要的问题是对各种键的描述[29].为了分析从Co-3 d到Ph-4 d,Ir-5d这些化合物的电子结构是如何演化的,我们计算了层间和面内磷族原子X(P,As)的键长以及层间键角和面内键角,如表2所示.1)面内键长大于层间键长显示TX4四面体沿z轴压缩; 2)面内键角依次减小,面间键角依次增大,在标准的正四面体中键角为109.28°,这种变化趋势充分说明,四面体形变的减小;3)TX以及层间X—X键长增加.在单质晶体中HCP结构的Co键长为2.51,面心立方(FCC)Rh中键长为2.69,面心立方(FCC)Ir晶体中键长为2.72和表2中列出的T—T键长差不多.所以这些化合物中存在很强的T—T成键.
表2 Ba T2As2和Ba T2P2中原子间的键长()和键角).其中表示面内磷族原子X间的键角,而表示相邻平面磷族原子X间的键角.为面内磷族原子间距,d为面间磷族原子间距
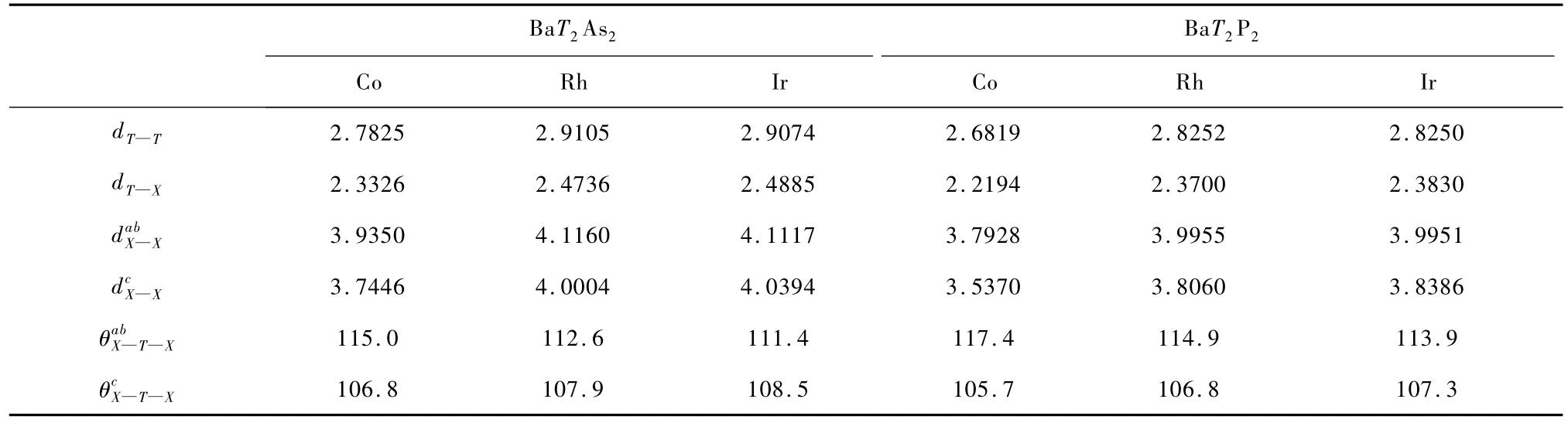
表2 Ba T2As2和Ba T2P2中原子间的键长()和键角).其中表示面内磷族原子X间的键角,而表示相邻平面磷族原子X间的键角.为面内磷族原子间距,d为面间磷族原子间距
Ba T2As2Ba T2P2Co Rh Ir Co Rh Ir dT—T2.7825 2.9105 2.9074 2.6819 2.8252 2.8250 dT—X2.3326 2.4736 2.4885 2.2194 2.3700 2.3830 d abX—X3.9350 4.1160 4.1117 3.7928 3.9955 3.9951 d cX—X3.7446 4.0004 4.0394 3.5370 3.8060 3.8386 θabX—T—X115.0 112.6 111.4 117.4 114.9 113.9 θcX—T—X106.8 107.9 108.5 105.7 106.8 107.3
BaCo2As2和BaCo2P2的每个自旋单胞态密度(DOS)和投影态密度(PDOS)如图2和图3所示.和以往的计算一样,态密度在费米面处有一个很高的尖峰,费米面处的态密度分别为3.87和3.64 eV-1/ Co.d-p杂化和d-d杂化很厉害,能带结构图4和图5中显示在费米能级处,布里渊区的A点附近以及M-G-Z高对称线处存在大量的平带结构,形成所谓的范霍夫畸点.我们分别作了往3z2-r2和xy轨道投影的能带结构,显示d-d:σ(xy轨道)的反键态是形成范霍夫畸点的主要因素.范霍夫畸点位于费米能级导致的高态密度很容易引起体系通过斯通纳机理往巡游铁磁态转变.
基于这点考虑,我们对铁磁态的BaCo2As2和BaCo2P2进行了计算,得到的磁矩分别为0.40和0.39μB/Co,相对于非磁态的能量分别减小了22和20 meV/f.u.,和以往的计算一致[21].如图6和图7所示自旋劈裂把范霍夫畸点从费米能级处移开,使得态密度在费米能及处形成一个双峰结构,费米能级正好处于双峰结构形成的谷底.使得体系稳定在铁磁态,位于费米能级之下的峰主要由d-d杂化的反键态形成,位于费米面之上的峰由d-p杂化的反键态形成.对于BaCo2P2如图2,从-6.2 eV到-4.4 eV主要是层间p-p杂化的成键态,从-4.4 eV到3.5 eV主要是Co的3d-3 d杂化以及Co和P之间杂化形成.对于BaCo2As2如图3从-5.6 eV到-4.1 eV主要是层间As-As的成键态,从-4.1 eV到3.4 eV主要是Co的3 d-3d杂化以及Co和As之间杂化形成.BaCo2As2和BaCo2P2的电子结构相似,但是BaCo2P2中d-p杂化的带宽要大一些,P的2 p轨道比As的3 p轨道的能级更低,这是由于在BaCo2P2中Co—P之间的键长要相对小一些,导致杂化更强.
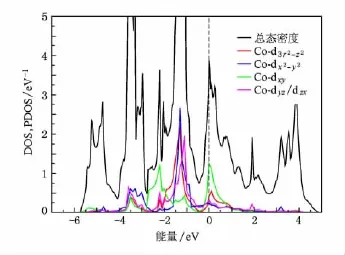
图2 非磁态BaCo2As2的总态密度及其在各个轨道的投影态密度

图3 非磁态BaCo2P2的总态密度及其在各个轨道的投影态密度
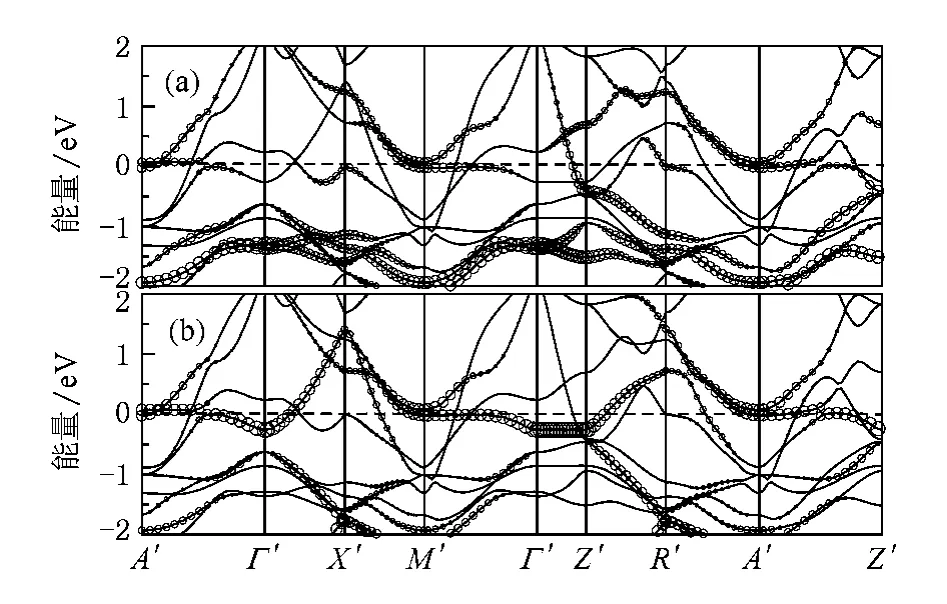
图4 非磁态BaCo2As2的能带结构,其中圆圈的大小表示投影到不同轨道的权重(a)往3 z2-r2轨道投影的能带结构; (b)往xy轨道投影的能带结构
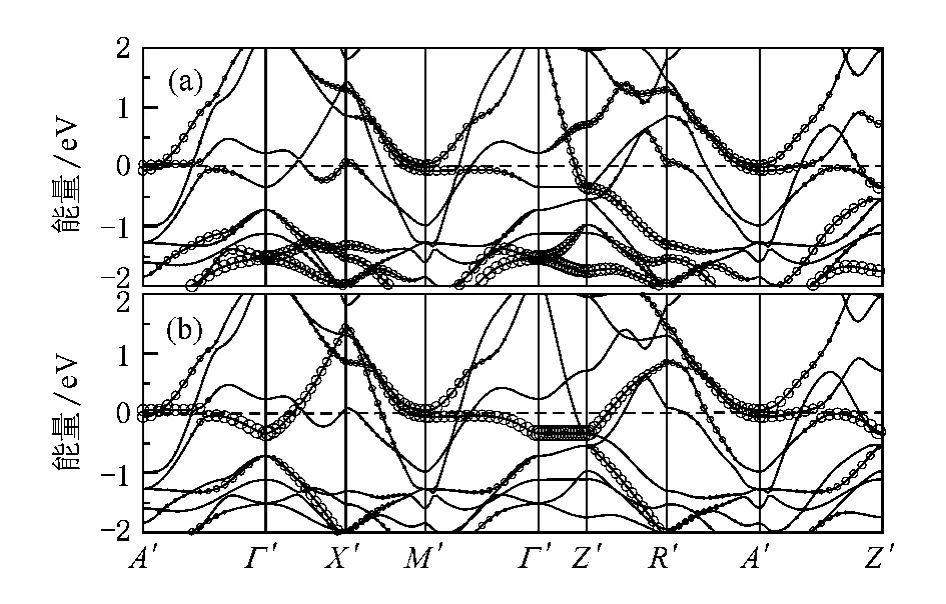
图5 非磁态BaCo2P2的能带结构,其中圆圈的大小表示投影到不同轨道的权重(a)往3 z2-r2轨道投影的能带结构;(b)往xy轨道投影的能带结构
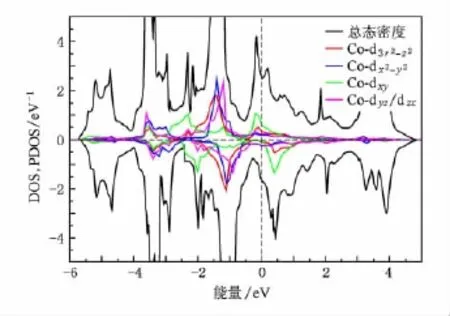
图6 铁磁态BaCo2As2的投影态密度
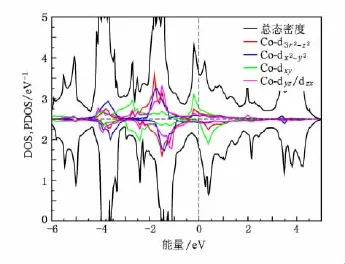
图7 铁磁态BaCo2P2的投影态密度
对于BaRh2As2和BaRh2P2如图8和图9所示,费米面处的态密度分别为2.07和2.02 eV-1/Co,由于Rh-4 d轨道比Co-3 d轨道更加扩展,由Rh的d-d反键态形成的平带结构被移往费米能级之上(位于0.3 eV),如图12(a)和(b)所示,导致费米能级处的态密度下降.对于BaRh2P2,如图9所示,从-6.5 eV到-3.6 eV主要是Rh—P的成键态,从-3.6 eV到4.6 eV主要是Rh的4 d-4 d杂化和Rh—P杂化的反键态形成,BaRh2As2的情况相似,但是带宽要小一些.

图8 非磁态BaRh2As2的投影态密度

图9 非磁态BaRh2P2的投影态密度
BaIr2As2和BaIr2P2的DOS和PDOS如图10和图11所示,费米面处的态密度分别为1.44和1.62 eV-1/Co,由于Ir-5 d轨道比Rh-4 d轨道更加扩展,由Ir的d-d反键态形成的平带结构被移往费米能级之上更高的能量处(位于0.8 eV),如图12(c)和(d)所示,导致费米能级处的态密度进一步下降,而且在费米能级处出现一个较小的峰,对应于实验上低温磁化率的异常[26].对于BaIr2P2从-7.8 eV到-4.1 eV主要是d-p杂化的成键态,-4.1 eV到4.5 eV主要是d-d杂化和d-p杂化的反键态组成.我们也计算了铁磁态的BaRh2X2和BaIr2X2,发现它们都收敛到非磁态,不同于BaCo2X2,下面我们会详细讨论其原因.

图10 非磁态BaIr2As2的投影态密度
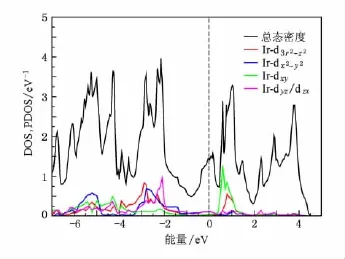
图11 非磁态BaIr2P2的投影态密度
这些化合物的电子结构有很多相似的地方: 1)都有很强的d-d成键,d-p成键也很强.Ba的s轨道都是远离费米能级之下(约-10.0 eV)所以Ba基本上都是2+离子态的,所有的键长都列在表2中.2)BaCo2X2中存在很强的面间磷族原子的成键.而在BaRh2X2和BaIr2X2中面间磷族原子成键要弱很多,这与表2中面间磷族原子键长从Co到Rh急剧增加相对应.3)在BaCo2X2中费米能级处的态密度要远大于BaRh2X2和BaIr2X2,这是由于Rh-4 d和Ir-5d电子要比Co-3d电子扩展许多,导致形成范霍夫畸点d-d杂化的反键态被移往远离费米能级之上的地方.如图12所示的能带结构中费米面处的态密度也相应的下降,斯通纳机理的消失,所以BaCo2X2会稳定在铁磁态,而BaRh2X2和BaIr2X2会稳定在非磁态.4)如图13和图14所示,BaCo2X2的费米面分别有3个,而且都是三维结构的,BaRh2X2和 BaIr2X2分别有2个费米面,从Co到Ir,位于Z点的费米面逐渐缩小直到消失,而且费米面逐渐变成的更接近于二维结构.5)在Ba T2P2中d-d成键和d-p成键的能带带宽要比Ba T2As2宽,这是由于Ba T2P2中键长要小.

图12 非磁态Ba T2X2的能带结构(a)非磁态BaRh2As2的能带结构;(b)非磁态BaRh2P2的能带结构;(c)非磁态BaIr2As2的能带结构;(d)非磁态BaIr2P2的能带结构
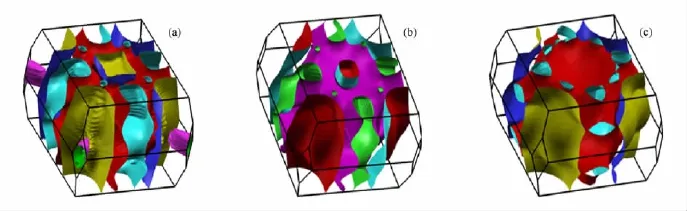
图13 非磁态Ba T2As2的费米面(a)BaCo2As2的费米面;(b)BaRh2As2的费米面;(c)BaIr2As2的费米面
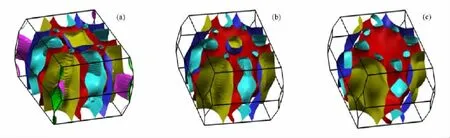
图14 非磁态Ba T2P2的费米面(a)BaCo2P2的费米面;(b)BaRh2P2的费米面;(c)BaIr2P2的费米面
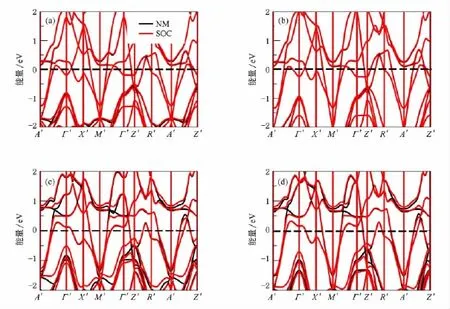
图15 (a)非磁态BaRh2As2的能带结构;(b)非磁态BaRh2P2的能带结构;(c)非磁态BaIr2As2的能带结构; (d)非磁态BaIr2P2的能带结构(黑色线条表示没有自旋轨道耦合,浅(红)色线条表示有自旋轨道耦合)
由于Rh和Ir都属于重元素,自旋轨道效应可能会对其电子结构有影响,基于这种考虑我们在计算中加入了自旋-轨道耦合相互作用,计算结果如图15所示.在BaRh2X2和BaIr2X2中,自旋-轨道耦合几乎不影响其能带结构,所以自旋轨道耦合作用几乎不影响其基本电子结构和性质.
4 .结论
我们计算了钴磷族化合物Ba T2P2和Ba T2As2(T=Co,Rh,Ir)的电子结构,并与实验和以往计算结果进行了对比.从Co到Rh,Ir的化合物中,我们对电子结构的演化做了详细的比较,发现由d-d之间的反键态形成的范霍夫畸点由于d轨道的扩展程度不同(从3d到5 d),随着原子数的增加从费米能处移往费米能级之上,导致费米能级处的态密度逐渐下降,从而在BaRh2X2和BaIr2X2中斯通纳机理逐步消失.所以BaCo2X2位于铁磁不稳定性的边缘,而BaRh2X2和BaIr2X2稳定在非磁态.计算表明自旋轨道耦合几乎对体系电子结构没有影响.
[1]Kamihara Y,Watanabe T,Hirano M,Hosono H 2008 J.Am.Chem.Soc.130 3296
[2]Chen G F,Li Z,Li G,Zhou J,Wu D,Dong J,Hu W Z,Zheng P,Chen Z J,Yuan H Q,Singleton J,Luo J L,Wang N L 2008 Phys.Rev.Lett.101 057007
[3]Zhu X,Yang H,Fang L,Mu G,Wen H H 20 0 8 Supercond. Sci.Technol.21 105001
[4]Rotter M,Tegel M,Johrendt D,Schellenberg I,Hermes W,Pottgen R 2008 Phys.Rev.B 78 020503(R)
[5]Rotter M,Tegel M,Johrendt D 2008 Phys.Rev.Lett.101 107006
[6]Wang X C,Liu Q Q,LüY X,Gao W B,Yang L X,Yu R C,Li F Y,Jin C Q 2008 Solid State Commun.148 538
[7]Hsu F C,Luo J Y,Yeh K W,Chen T K,Huang T W,Wu P M,Lee Y C,Huang Y L,Chu Y Y,Yan D C,Wu M K 2008 Proc.Natl.Acad.Sci.USA 105 14262
[8]Liu S,Li B,Wang W,Wang J,Liu M 2010 Acta Phys.Sin.59 4245(in Chinese)[刘甦、李斌、王玮、汪军、刘楣2010物理学报59 4245]
[9]Yang S,You W L,Gu S J,Lin H Q 2009 Chin.Phys.B 18 2545
[10]Li Z C,Zhou F 2010 Chin.Phys.B 19
[11]Hamlin J J,Baumbach R E,Zocco D A,Sayles T A,Maple M B 2008 J.Phys.Condens.Matter 20 365220
[12]Dong J,Zhang H J,Xu G,Li Z,Li G,Hu W Z,Wu D,Chen G F,Dai X,Luo J L,Fang Z,Wang N L 2008 Europhys.Lett.83 27006
[13]Cruz C de la,Huang Q,Lynn J W,Li J Y,Ratcliff W,Zarestky J L,Mook H A,Chen G F,Luo J L,Wang N L,Dai P C 2008 Nature 453 899
[14]Kofu M,Qiu Y,Bao W,Lee S H,Chang S,Wu T,Wu G,Chen X H 2009 New J.Phys.11 055001
[15]Takumi W,Hiroshi Y,Toshio K,Yoichi K,Hidenori H,Masahiro H,Hideo H 2007 Inorg.Chem.46 7719
[16]Takumi W,Hiroshi Y,Yoichi K,Toshio K,Masahiro H,Hideo H 2008 J.Solid State Chem.181 2117
[17]Li Z,Chen G F,Dong J,Li G,Hu W Z,Wu D,Su S,Zheng P,Xiang T,Wang N L,Luo J L 2008 Phys.Rev.B 78 R060504
[18]Ronning F,Kurita N,Bauer E D,Scott B L,Park T,Klimczuk T,Movshovich R,Thompson J D 2008 J.Phys.Condens.Matter.20 342203
[19]Takashi M,Hiroshi Y,Toshio K,Yoichi K,Masahiro H,Hideo H 2008 Solid State Commun.147 111
[20]Hiroshi Y,Ryuto K,Toshio K,Yoichi K,Masahiro H,Tetsuya N,Hitoshi O,Hideo H 2008 Phys.Rev.B 77 224431
[21]Sefat A S,Singh D J,Jin R,McGuire M A,Sales B C,Mandrus D 2009 Phys.Rev.B 79 024512
[22]Yogesh S,Lee Y,Nandi S,Kreyssig A,Ellern A,Das S,Nath R,Harmon B N,Goldman A I,Johnston D C 2008 Phys.Rev.B 78 104512
[23]Daigorou H,Tomohiro T,Ryuji H,Hiroko A K,Hidenori T 2009 J.Phys.Soc.Jpn.78 023706
[24]Berry N,Capan C,Seyfarth G,Bianchi A D,Ziller J,Fisk Z 2009 Phys.Rev.B 79 180502(R)
[25]Fang Z,Terakura K 2002 J.Phys.Condens.Matter.14 3001
[26]Perdew J,Burke K,Ernzerhof M 1996 Phys.Rev.Lett.77 3865
[27]Wang G T,Qian Y M,Xu G,Dai X,Fang Z 2010 Phys.Rev.Lett.104 047002
[28]Shein I R,Ivanovskii A L 2009 JETP Lett.89 357
[29]Johrendt D,Felser C,Jepsen O,Andersen O K,Mewis A,Rouxel J 1997 J.Solid State Chem.130 254
PACS:71.20.-b,71.18.+y,75.20.-g
Electronic structures cobalt group pnictides:Ba T2P2and Ba T2As2(T=Co,Rh,Ir)*
Qian Yu-MinXu Gang
(Beijing National Laboratory for Condensed Matter Physics,Institute of Physics,Chinese Academy of Sciences,Beijing 100190,China)
(Received 12 July 2010;revised manuscript received 9 September 2010)
We study the electronic structure of cobalt pnictides:Ba T2P2and Ba T2As2(T=Co,Rh,Ir)by the density functional theory within generalized gradient approximation,and find that the ferromagnetisms in BaCo2As2and BaCo2P2are due mostly to the high density of states(Van-Hove singularity)at Fermi level,which induce Stoner ferromagnetism.In these compounds,from Co-3 d to Rh-4d,then to Ir-5d,the d-d bonding and the d-p bonding in the TX4(X=P,As) layers are strengthened.As a result,the antibonding d-d states are pushed away from the Fermi level,and the ferromagnetisms are suppressed in Rh and Ir compounds.The evolutions and the detailed electronic structures of these compounds are studied and compared,and spin orbital coupling interaction is negligible.
electronic structure,Van-Hove singularity,Stoner instability
*国家自然科学基金(批准号:90921004)资助的课题.
E-mail:yuminqian@gmail.com
*Project supported by the National Natural Science Foundation of China(Grant No.90921004).
E-mail:yuminqian@gmail.com
猜你喜欢
数学物理学报(2022年5期)2022-10-09
中外文摘(2021年7期)2021-04-23
发明与创新(2020年39期)2020-10-15
数学物理学报(2019年6期)2020-01-13
发明与创新·小学生(2019年12期)2019-12-05
陕西中医(2018年6期)2018-08-29
黑龙江电力(2017年1期)2017-05-17
中国塑料(2016年1期)2016-05-17
学生天地·小学低年级版(2016年9期)2016-05-14
读写算·教研版(2016年8期)2016-05-07
