
基因组尺度代谢网络自动重构及分析工具研究进展
2012-09-29 07:25郝彤马红武赵学明
生物工程学报 2012年6期
郝彤,马红武,赵学明
1 天津大学化工学院生物工程系,天津 300072
2 教育部系统生物工程重点实验室,天津 300072
3 天津大学-爱丁堡大学系统生物学与合成生物学联合研究中心,天津 300072
基因组尺度代谢网络自动重构及分析工具研究进展
郝彤1,2,3,马红武1,2,3,赵学明1,2,3
1 天津大学化工学院生物工程系,天津 300072
2 教育部系统生物工程重点实验室,天津 300072
3 天津大学-爱丁堡大学系统生物学与合成生物学联合研究中心,天津 300072
高通量数据的产出为基因组尺度代谢网络的构建提供了基础,但同时也对网络构建和分析方法的改进提出了挑战。随着数据量的不断增大,耗时耗力的人工构建及分析已经无法满足模型发展的需要,因而各种自动化的方法应运而生。模型构建和分析的自动化不仅能够大幅度提高模型构建和解析的速度,同时对于模型构建和分析方法的标准化和程序化也有着不可替代的作用。文中结合作者的实际研究经验,对基因组尺度代谢网络构建的自动化进程和主要的代谢网络分析工具进行了较为详细的介绍,总结了代谢网络自动重构的流程,并提出了目前面对的主要问题和未来的研究方向。
基因组尺度,代谢网络,自动重构,网络分析
Abstract:High-throughput data supply a basis for the reconstruction of genome-scale metabolic networks, andmeanwhile bring challenges to the reconstruction and analysis methods. With the increasing of data quantity, the time-consuming manual reconstruction and analysis are far behind the improvement of models. Therefore, various automatic methods emerge. The automatic reconstruction and analysis have irreplaceable effect in the standardization and programming of reconstruction and analysis methods, as well as largely improving the speed of reconstruction and understanding of the metabolic network. In this review, we introduced the progress of automatic reconstruction and the main analysis tools of genome-scale metabolic network. We further summarized the workflow of automatic reconstruction.The difficulties and perspectives on this research field are also discussed.
Keywords:genome-scale, metabolic network, automatic reconstruction, network analysis
近些年来,随着以基因组测序为代表的大规模数据产出,传统的生物学研究方式正在发生改变,在基因组测序和注释海量数据的基础上,基因组尺度的代谢网络重构迅速发展起来[1-2]。基因组尺度代谢网络已经成为研究生物代谢系统不可缺少的工具[3-4],在设计代谢工程经典途径、代谢物合成、代谢通量分析、不同物种代谢途径之间的进化分析、挖掘组学数据信息以及选择酶工程靶标物方面都具有重要的应用[5-6]。基因组尺度代谢网络模型作为工具,无论在生物体以及生命活动的理论研究上,还是在指导代谢工程进行工程菌改造上,都具有非常重要的理论和实践意义。
随着基因组测序技术的提高,全基因组测序生物的数量呈指数逐年增长,截止至2011年10月,已经有1 943个物种全基因组测序完成 (其中细菌1 673株,古细菌119株,真核生物151株),另外,1 028个物种的全基因组草图也已经完成并已收录在 GOLD数据库 (GOLD:http://www.genomesonline.org/cgi-bin/GOLD/bin/gold.cgi. [2011-10-31]) 中。这些数据构成了庞大的信息资源,为研究生物体的生理特征、代谢表型、病理分析提供了基础。正是由于数据量的庞大规模和快速发展,生物学研究对于计算机方法处理数据的要求也越来越高[2]。理论上来说,有多少物种的全基因组测序完成,就应该存在多少个对应的基因组尺度代谢网络,然而截止至2011年10月的统计,目前只有81个物种的126个基因组尺度代谢网络重构完成,并收录在基因组尺度代谢网络模型数据库中 (http://synbio.tju.edu.cn/GSMNDB/gsmndb.htm),基因组尺度代谢网络数量远远小于已测序物种的数量 (图1)。造成这种情况的原因主要有 2个:1) 我们对很多物种的生理生化机制了解有限;2) 重构过程中需要大量耗时耗力的人工修正工作[6-7]。前者需要通过长时间生物实验研究逐步改善,而后者则需要通过提高重构过程的自动化水平以减少人工修正过程来解决。同时网络重构的速度迅速提高,能够为我们提供更多了解生物生理生化机制的素材,对第一个问题的解决也具有重要的推动作用。近些年有一些致力于通过计算机平台自动化重构过程的研究[8-11],这些研究已经取得了一些成绩,但是其中仍然需要大量的人工修正工作[12]。另一方面,代谢网络分析工具的开发对于代谢网络的发展和应用也起到了重要的推动作用,因此,代谢网络重构的自动化和分析工具的开发成为提高网络重构速度并推动代谢网络研究发展的重要问题,逐渐引起研究者们的兴趣。在这里,作者结合自己的研究经历,较为详细地阐述基因组尺度代谢网络自动化重构的研究进展、介绍主要的代谢网络分析工具,并总结自动化重构的流程。

图1 已测序物种和已构建基因组尺度代谢网络数目Fig. 1 The number of sequenced species and reconstructed genome-scale metabolic networks.
1 基因组尺度代谢网络自动重构进展
在网络重构方面,Palsson实验组是走在最前面的,1999年该实验组重构了第一个基因组尺度代谢网络模型[13],此后又先后重构了包括人类和微生物在内的十几个物种的基因组尺度模型,有些物种的模型经过不断修正,先后重构了多个版本,重构方法也不断改进。以大肠杆菌Escherichia coli为例,该实验室最初建立的大肠杆菌基因组尺度代谢网络模型iJE660[14]包含660个基因,以数据库和文献信息为基础,通过大量的人工修正辅助建立,并通过通量平衡分析(Flux balance analysis,FBA) 方法进行了生物量合成以及必需基因模拟,其中必需基因预测的准确率为 86%。第二代模型 iJR904[15]利用基因组信息、生化和生理学数据对iJE660进行了扩充,并进行了元素和电荷守恒的修正,iJR904在限氧并以α-酮戊二酸为底物的条件下,对生物量合成的预测准确性显著高于iJE660,并且必需基因预测的准确率达到88%。在iJR904的基础上,他们又根据大肠杆菌的基因组功能注释、EcoCyc数据库以及文献信息对网络进行了再次扩展和修正,建立了iAF1260模型[16],热力学信息的加入使网络包含的信息更加完善,该模型对必需基因预测的准确率进一步提高到了 92%。通过对iAF1260的进一步完善,2011年该组构建了大肠杆菌iJO1366模型[17],该模型在iAF1260的基础上添加了106个新的基因,虽然该模型在底物利用和基因必需性预测方面比 iAF1260的准确性略低,但是包含了更加广泛的代谢网络信息。2010年Palsson发表的综述文章中系统地介绍了重构一个高质量代谢网络的96个步骤[18],这些步骤在COBRA的自动分析基础上辅以大量人工修正工作,特别是运输反应的添加,由于文献信息的缺乏,很难实现自动化。根据这样的步骤和方法,对于不同的物种,重构一个全基因组尺度代谢网络的时间在6个月到2年不等。
可见,随着构建方法的发展,代谢网络模型的规模正在不断扩大,人工构建已经无法满足模型发展的需要,研究者对于自动化模型构建的要求也越来越高。本文从网络构建的几个步骤分别介绍自动化网络构建的进展。
1.1 自动化初步重构
从自动化程度方面来说,在代谢网络的重构过程中,自动化程度较高的是原始数据收集步骤。以人类代谢网络为例,从KEGG和Reactome数据库中能够直接下载人类代谢网络中包含的反应及途径信息,通过与这些数据库关联,可以得到人类代谢网络重构的初始数据。基于此,Schwarz等[19]开发了YANAsquare软件,该软件可以通过对 KEGG数据库的自动搜索初步重构代谢网络。马红武等也已经开发了能够自动从KEGG数据库提取各物种代谢网络数据的网络工具 (http://csb.inf.ed.ac.uk/kneva/)。此外,孙际宾等[20]开发的 IdentiCS软件可以从基因组测序信息对基因组进行注释,并进行网络的初步重构,对于测序时基因组覆盖度较低的物种,该软件在网络初步重构方面具有优势。直接由基因组或数据库信息重构的网络可以用于网络特征和拓扑结构分析,但是这样的网络比较粗糙,还不能满足代谢通量分析的要求,需要进一步的修正和完善[21]。
为了得到尽可能完善的数据信息以提高重构网络的质量,一些综合多个数据源、自动获取信息初步重构网络的软件及软件平台发展起来。DeJongh等[22]开发了用于网络自动初步重构的SEED平台,该平台将所有基因组信息 (不依赖于物种) 按照功能分为若干个子系统,每个子系统中都包含各种微生物的基因注释和功能信息,以及不同微生物之间的基因功能差异,重构时从各个子系统中提取出所需微生物的基因组信息汇总起来完成重构,通过与人工修正的 E. coli iJR904、H. pylori iIT341以及 Oliverira等[23]重构的乳酸乳球菌Lactococcus lactis模型比较,发现用SEED重构的模型可以分别覆盖到已重构模型中87%、90%和83%的反应。
Arakawa等[24]开发的GEM System软件从基因组注释出发自动构建代谢网络,并建立计量学矩阵,该模型可以覆盖KEGG和EcoCyc数据库中大肠杆菌100%和92.8%的数据,并覆盖E. coli iJR904中95.06%的数据。
Cottret等[25]开发了基于代谢物组学的基因组尺度代谢网络构建工具MetExplore、该工具建立在50个物种数据集的基础上,从代谢物角度出发,将整个代谢网络表示为一个无重复代谢物大网络图,而不是区分为多个包含重复代谢物的代谢途径,从而更加清晰地描述了代谢物之间的关系。
上述软件和工具的出现一定程度上减少了代谢网络模型中的人工工作量,从而提高了模型的构建速度。然而虽然SEED、GEM等方法已经能够很大程度上覆盖一些模式菌已构建代谢网络中的数据,但仍然无法替代很多重构代谢网络中的人工修正工作。这些自动构建的网络中大多不包含大部分与运输相关的反应以及没有对应基因的反应,而且在处理酶号不全的酶时容易出现错误,其重构网络的质量无法满足直接用于高质量分析预测的需要。因而,直接由基因组或数据库信息得到的重构网络需进行进一步的修正。
1.2 自动化网络空白填补
网络的结构分析是查找网络空白的一种有效方式,Arakawa等[24]开发的GEM System软件在从基因组注释出发初步重构的网络基础上,通过途径连通性分析自动填补了一些网络空白。本文作者等[26-27]在对人类代谢网络进行扩展的工作中,将代谢网络转化为反应图,通过对反应图中最小弱连接体的连通性分析确定了网络中的空白。
除了结构分析的方法外,优化算法也可以用在网络空白的自动填补中,在小鼠代谢网络模型的构建中,Palsson实验组[28]采用了SMILEY方法来辅助填补网络中的空白,该方法在保证菌体正常生长的条件下,通过线性规划自动确定用于填补空白的候选反应,再通过人工查证的方法来确定最终的解决方案[29]。Costas D Maranas实验组[30]提出了通过查找末端代谢物查找和填补网络空白的方法 GapFind和 GapFill,末端代谢物是指在其参与的所有反应中都只作为底物或只作为产物的物质,GapFind方法通过代谢末端的计算找到网络中的末端代谢物,将之确定为网络中的空白,在此基础上,GapFill方法以网络中的所有反应以及 MetaCyc数据库中的所有反应为数据基础,进行线性规划的计算,确定能够用于填补空白的反应,或通过对反应可逆性的修改消除部分网络空白。该方法填补了网络中与末端代谢物相关的网络空白,并且添加了一部分与网络空白相关的运输反应。
1.3 自动化模拟修正
代谢网络构建的目的是用于模拟生物的生理表型和代谢过程,因而重构模型在模拟方面需要有较高的准确率。为了满足这一要求,在模型重构过程中,模拟修正成为重要的一步。该步骤的目的是通过“模拟-与实验比较-修正”的过程保证重构模型对于已有实验结果的预测准确率,从而提高对于未知实验预测结果的可信度。
模拟修正要求模拟与修正能够互相结合,目前许多软件或工具包 (如 CellNetAnalyzer[31]、SBRT[32]、OptFlux[33]等) 都可以进行基于通量平衡分析的模拟计算,但是当模拟结果与实验数据不符时,对网络的修正还需通过人工查阅文献完成。为了探索这个环节的自动化问题,Costas D Maranas实验组[34]开发了GrowMatch方法,用于自动修正单基因敲除实验中,模型模拟与实验结果不符的情况。该方法通过最优化确定使模拟结果与实验结果一致需改变的反应数的最小值,随后对两种不同的情况GNG (模拟结果为生长,实验结果为不生长) 和NGG (模拟结果为不生长,实验结果为生长) 采用不同的策略进行修正,对于 GNG,通过抑制特定反应达到抑制生长的目的,对于 NGG,则通过添加适当的途径完善网络功能,从而使模型能够模拟菌种的生长。通过将该方法应用于大肠杆菌iAF1260模型,修正了56/72个GNG以及13/38个NGG,从而使模型单基因敲除的预测准确率由 90.6%提高到94.6%。该方法在枯草芽胞杆菌模型构建中的应用使模型对单基因敲除的准确率由 89.7%提高到 93.1%[35]。
另外,Palsson实验组开发的商业化网络重构软件 SimphenyTM融合了基因组序列,基因表达,蛋白组学和代谢物组学的数据,并包含了多种计算功能,可用于基因组尺度代谢网络的自动重构和修正。
2 基因组尺度代谢网络分析工具
随着重构基因组尺度代谢网络数量的不断增加,人们对于网络自动分析的研究也逐渐深入。代谢网络分析工具从最初的利用 LINDO等数学分析工具的线性规划工具包[13-14],到现在已有很多专门针对生物代谢网络的软件和工具包开发出来。这里对主要的单机软件和网络软件进行介绍。
2.1 单机软件
虽然代谢网络的重构和分析是两个独立的部分,但是一些网络平台和软件同时具备了这两方面的功能。Simpheny软件在网络构建之外,同时具备底物利用、产物生成、基因敲除、适应性进化分析等多种分析功能。YANAsquare软件也能够实现代谢网络的可视化和分析。另外还有一些专门用于网络分析的工具。Palsson实验组[36]开发的免费网络分析工具包 COBRA在 Matlab环境下运行,使用基于约束的方法对基因组尺度的网络模型进行细胞表型的量化预测,它包含了多种 FBA算法,能够完成通量分布计算、不同底物生长表型预测、基因敲除分析、通量可变性分析 (系统鲁棒性分析、表型相平面分析)、前体必要性分析、副产物产量计算等多种分析功能,2011年开发的新版本COBRA Toolbox v2.0[37]又增加了空白填补、13C分析、组学分析和可视化等功能。
Sung Yup Lee实验组[38]开发了MetaFluxNet软件作为代谢网络数据管理的平台对代谢网络进行可视化和简单的通量平衡分析。Pabinger等[39]开发的 MEMOSys平台能够用于数据的管理、存储和扩展,从而为新模型的构建提供有用的信息。目前该平台包含6个已注释的微生物代谢网络模型。更值得注意的是,这个平台包含了模型比较的功能,能够比较模型之间反应、代谢物和基因的不同之处。另外,FluxAnalyzer工具包[31]能够进行网络的拓扑结构分析、代谢通量分析、基元模式分析和端途径分析等,同时能够通过交互和可视化的方式综合分析代谢网络和信号调控网络。iMAT工具包[40]能够融合转录组学和蛋白组学的数据预测基因组尺度代谢网络模型中酶的代谢通量。SBRT软件[32]可用于分析计量学网络,并从其他领域如图论、几何学、组合学等借鉴了一些方法。OptFlux软件平台[33]首次应用了菌种优化算法,即能够识别代谢工程目标,对代谢模型进行野生菌和突变菌的表型模拟。FASIMU软件[41]包含了使用热力学作为限制条件的通量平衡分析方法,并且在目标函数和约束条件的选择方面更加灵活,用户可以根据需要选择合适的目标函数和约束条件。
2.2 网络软件
除了单机版的分析软件,一些基于网络的工具也被开发出来,相对于单机软件,网络工具具有更加方便的应用性和可拓展性。Beste等[42]发表了第一个基于互联网的基因组尺度模型GSMN-TB,该模型在 Linux 操作系统下运行,可用于结核分枝杆菌Mycobacterium tuberculosis的底物消耗速率的计算、基因必需性预测以及通量可变性分析。Le Fèvre等[43]开发了CycSim网络工具,该工具支持基因敲除模拟、预测单或多基因敲除突变体的生长表型,而且可以得到直观的可视化代谢图。该工具内部的模型库包含酿酒酵母Saccharomyces cerevisae、E. coli和不动杆菌Acinetobacter baylyi ADP1这3个菌的模型数据、培养基组分数据及其他生化数据。除了这3个菌型之外,该软件目前还无法分析其他物种的模型文件。
Cvijovic等[44]开发的 BioMet网络工具包可用于高通量数据的分析,该工具包分为3个部分:BioOpt,Reporter Features和 Reporter Subnetwork。BioOpt用于模型的通量平衡分析;Reporter Features可用于分析生物网络的拓扑结构,通过拓扑结构分析可以确定生物网络的一些网络特征;Reporter Subnetwork用于识别发生直接或间接扰动时发生变化的代谢子网络。同时,该工具箱还包含了Saccharomyces cerevisiae,天蓝色链霉菌Streptomyces coelicolor、Lactococcus lactis、黑曲霉 Aspergillus niger、构巢曲霉 Aspergillus nidulans、米曲霉Aspergillus oryzae和谷氨酸棒杆菌Corynebacterium glutamicum的代谢网络数据。
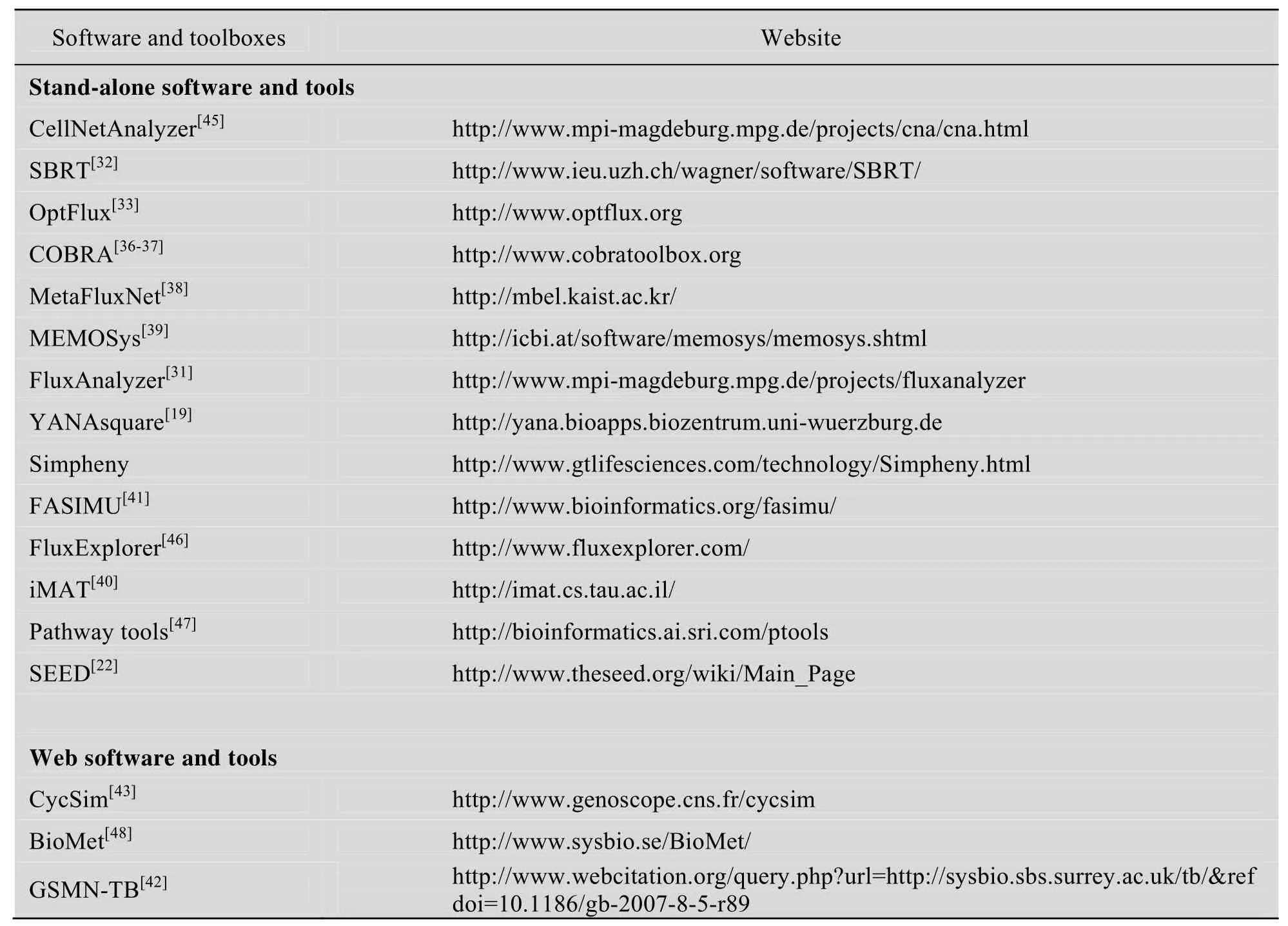
表1 常用代谢网络分析软件和工具包Table 1 Software and toolboxes for metabolic network analysis
软件及工具包的使用使基因组尺度代谢网络的分析更加简单易学,应用范围更加广泛,在生物预测及生物代谢机理研究方面也具有了更深入探索的可能性。常用代谢网络分析软件及工具包及其对应的网址如表1所示。
3 基因组尺度代谢网络自动重构流程总结及展望
综合上文所述,基因组尺度代谢网络自动重构的流程主要分为初步重构、空白填补及模拟修正3个部分,其中初步重构的自动化是目前发展比较成熟的部分。对于高质量代谢网络的构建,仅从单一数据库获得信息是不够的,构建者可以选择建立在多个数据库基础上的自动重构软件,如文中提到的SEED、GEM System,来得到初步重构的网络。在空白填补的过程中,如果根据结构来填补,可以采用gapfind、gapfill方法以及最小弱连接体连通性分析的方法,如果根据通量平衡分析来填补,可以采用SMILEY算法,如果初步重构网络是利用GEM System软件获得,继续利用该软件的功能进行空白填补则更加快捷。在空白填补的过程中,出现一个网络空白有多个候选反应进行填补时,需要进行必要的人工选择。自动模拟修正方面的自动化方法目前研究较少,利用 GrowMatch方法可以对网络进行定性的修正,该方法已用于利用基因必要性数据进行大肠杆菌和枯草芽胞杆菌网络的修正。对于定量的模拟修正,目前仍需根据模拟分析软件得到的结果,通过人工分析进行。在代谢网络分析方面,目前开发出的软件很多,使用者可以根据研究目的和需求选择具有相关功能的软件,本文介绍的大多数软件都具有基本的通量平衡分析功能。
基因组尺度代谢网络自动化重构及分析是基因组尺度代谢网络研究的发展趋势,虽然自动化过程不可能完全取代人工判断在代谢网络构建和分析中的作用,但是其规范、快速的特点对于促进代谢网络的发展和应用仍然起着不可替代的作用。研究者们对其进行的研究得到了许多具有指导意义的方法和策略。但是,目前自动化重构的网络质量仍然不高,基于软件的网络分析也需要大量人工操作的辅助,目前的研究存在以下主要问题。
首先,数据收集的全面性有待提高。数据是网络构建和网络修正的基础,仅靠数据库和基因组注释得来的信息往往并不全面,为了提高网络的质量,需要从更加广泛的数据源得到能够用于完善代谢网络的数据。
其次,在网络空白查找过程中,目前的自动化方法仅考虑了与末端代谢物相关的网络空白,对于非末端代谢物的考虑不足,通量分析的方法在空白填补的过程中还不能做到完全的自动化,根据不同情况需人工进行不同的处理和修正,在这一方面,文献信息的匮乏,特别是与运输反应相关的实验研究的缺乏是网络空白填补中的重要问题。
再次,目前还没有一个软件能够实现高水平基因组尺度代谢网络整个重构过程的自动化,网络构建中需人工工作最多的地方是模拟修正,除了必需基因预测,对于其他方面,当模拟分析结果与实验结果不一致时,尚缺乏自动修正的方法。
最后,在网络分析过程中,自动化软件对计算结果生物学意义的分析和判断仍然缺乏统一的标准,另外,不同分析软件对于输入文件的格式要求也各有不同,使得利用多个软件进行的代谢网络分析过程增加了许多数据格式转换的工作。
随着海量数据的不断产生以及自动化水平的提高,模型构建和分析的过程也将逐步走向标准化和程序化,这一过程在促进代谢网络模型本身发展的同时,对于其他网络,如转录调控网络,信号转导网络等的构建也必定大有裨益。
REFERENCES
[1] Notebaart RA, van Enckevort FH, Francke C, et al.Accelerating the reconstruction of genome-scale metabolic networks. BMC Bioinform, 2006, 7: 296.
[2] Francke C, Siezen RJ, Teusink B. Reconstructing the metabolic network of a bacterium from its genome. Trends Microbiol, 2005, 13(11): 550−558.
[3] Feist AM, Palsson BØ. The growing scope of applications of genome-scale metabolic reconstructions using Escherichia coli. Nat Biotechnol, 2008, 26(6): 659−667.
[4] Pál C, Papp B, Lercher MJ, et al. Chance and necessity in the evolution of minimal metabolic networks. Nature, 2006, 440(7084): 667−670.
[5] Soh KC, Hatzimanikatis V. DREAMS of metabolism. Trends Biotechnol, 2010, 28(10):501−508.
[6] Wang H, Ma HW, Zhao XM. Progress in genome-scale metabolic network: a review. Chin J Biotech, 2010, 26(10): 1340−1348.
王晖, 马红武, 赵学明. 基因组尺度代谢网络研究进展. 生物工程学报, 2010, 26(10): 1340−1348.
[7] Palsson B. Metabolic systems biology. FEBS Lett,2009, 583(24): 3900−3904.
[8] Feist AM, Herrgård MJ, Thiele I, et al.Reconstruction of biochemical networks in microorganisms. Nat Rev Microbiol, 2009, 7(2):129−143.
[9] Poolman MG, Bonde BK, Gevorgyan A, et al.Challenges to be faced in the reconstruction of metabolic networks from public databases. Syst Biol (Stevenage), 2006, 153(5): 379−384.
[10] Gevorgyan A, Poolman MG, Fell DA. Detection of stoichiometric inconsistencies in biomolecular models. Bioinformatics, 2008, 24(19): 2245−2251.
[11] Herrgård MJ, Fong SS, Palsson BØ. Identification of genome-scale metabolic network models using experimentally measured flux profiles. PLoS Comp Biol, 2006, 2(7): e72.
[12] Ruppin E, Papin JA, de Figueiredo LF, et al.Metabolic reconstruction, constraint-based analysis and game theory to probe genome-scale metabolic networks. Curr Opin Biotechnol, 2010, 21(4):502−510.
[13] Edwards JS, Palsson BO. Systems properties of the Haemophilus influenzae Rd metabolic genotype. J Biol Chem, 1999, 274(25): 17410−17416.
[14] Edwards JS, Palsson BO. The Escherichia coli MG1655 in silico metabolic genotype: its definition, characteristics, and capabilities. Proc Natl Acad Sci USA, 2000, 97(10): 5528−5533.
[15] Reed JL, Vo TD, Schilling CH, et al. An expanded genome-scale model of Escherichia coli K-12(iJR904 GSM/GPR). Genome Biol, 2003, 4(9): R54.
[16] Feist AM, Henry CS, Reed JL, et al. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol Syst Biol, 2007, 3: 121.
[17] Orth JD, Conrad TM, Na J, et al. A comprehensive genome-scale reconstruction of escherichia coli metabolism-2011. Mol Syst Biol, 2011, 7: 535.
[18] Thiele I, Palsson BØ. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc, 2010, 5(1): 93−121.
[19] Schwarz R, Liang CG, Kaleta C, et al. Integrated network reconstruction, visualization and analysis using YANAsquare. BMC Bioinform, 2007, 8: 313.
[20] Sun JB, Zeng AP. IdentiCS-identification of coding sequence and in silico reconstruction of the metabolic network directly from unannotated low-coverage bacterial genome sequence. BMC Bioinform, 2004, 5: 112.
[21] Durot M, Bourguignon PY, Schachter V.Genome-scale models of bacterial metabolism:reconstruction and applications. FEMS Microbiol Rev, 2009, 33(1): 164−190.
[22] DeJongh M, Formsma K, Boillot P, et al. Toward the automated generation of genome-scale metabolic networks in the SEED. BMC Bioinform,2007, 8: 139.
[23] Oliveira AP, Nielsen J, Förster J. Modeling lactococcus lactis using a genome-scale flux model.BMC Microbiol, 2005, 5: 39.
[24] Arakawa K, Yamada Y, Shinoda K, et al. GEM System: automatic prototyping of cell-wide metabolic pathway models from genomes. BMC Bioinform, 2006, 7: 168.
[25] Cottret L, Wildridge D, Vinson F, et al.Metexplore: a web server to link metabolomic experiments and genome-scale metabolic networks.Nucleic Acids Res, 2010, 38(Web Server issue):W132−W137.
[26] Hao T, Ma HW, Zhao XM, et al.Compartmentalization of the edinburgh human metabolic network. BMC Bioinform, 2010, 11(1):393.
[27] Hao T, Ma HW, Zhao XM, et al. The reconstruction and analysis of tissue specific human metabolic networks. Mol Biosyst, 2012,8(2): 663−670.
[28] Reed JL, Patel TR, Chen KH, et al. Systems approach to refining genome annotation. Proc Natl Acad Sci USA, 2006, 103(46): 17480−17484.
[29] Sigurdsson MI, Jamshidi N, Steingrimsson E, et al.A detailed genome-wide reconstruction of mouse metabolism based on human Recon 1. BMC Syst Biol, 2010, 4(1): 140.
[30] Satish Kumar V, Dasika MS, Maranas CD.Optimization based automated curation of metabolic reconstructions. BMC Bioinform, 2007, 8: 212.
[31] Klamt S, Stelling J, Ginkel M, et al. FluxAnalyzer:exploring structure, pathways, and flux distributions in metabolic networks on interactive flux maps. Bioinformatics, 2002, 19(2): 261−269.
[32] Wright J, Wagner A. The systems biology research tool: evolvable open-source software. BMC Syst Biol, 2008, 2(1): 55.
[33] Rocha I, Maia P, Evangelista P, et al. OptFlux: an open-source software platform for in silico metabolic engineering. BMC Syst Biol, 2010, 4(1): 45.
[34] Kumar VS, Maranas CD. GrowMatch: an automated method for reconciling in silico/in vivo growth predictions. PLoS Comput Biol, 2009, 5(3):e1000308.
[35] Henry CS, Zinner JF, Cohoon MP, et al. iBsu1103:a new genome-scale metabolic model of Bacillus subtilis based on SEED annotations. Genome Biol,2009, 10(6): R69.
[36] Becker SA, Feist AM, Mo ML, et al. Quantitative prediction of cellular metabolism with constraintbased models: the COBRA Toolbox. Nat Protoc,2007, 2(3): 727−738.
[37] Schellenberger J, Que R, Fleming RM, et al.Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat Protoc, 2011, 6(9): 1290−1307.
[38] Lee DY, Yun H, Park S, et al. MetaFluxNet: the management of metabolic reaction information and quantitative metabolic flux analysis.Bioinformatics, 2003, 19(16): 2144−2146.
[39] Pabinger S, Rader R, Agren R, et al. MEMOSys:bioinformatics platform for genome-scale metabolic models. BMC Syst Biol, 2011, 5(1): 20.
[40] Zur H, Ruppin E, Shlomi T. iMAT: an integrative metabolic analysis tool. Bioinformatics, 2010,26(24): 3140−3142.
[41] Hoppe A, Hoffmann S, Gerasch A, et al. FASIMU:flexible software for flux-balance computation series in large metabolic networks. BMC Bioinform, 2011, 12(1): 28.
[42] Beste DJ, Hooper T, Stewart G, et al. GSMN-TB: a web-based genome-scale network model of mycobacterium tuberculosis metabolism. Genome Biol, 2007, 8(5): R89.
[43] Le Fèvre F, Smidtas S, Combe C, et al. Cycsim-an online tool for exploring and experimenting with genome-scale metabolic models. Bioinformatics,2009, 25(15): 1987−1988.
[44] Cvijovic M, Olivares-Hernández R, Agren R, et al.BioMet toolbox: genome-wide analysis of metabolism. Nucleic Acids Res, 2010, 38(Web Server): W144−W149.
[45] Klamt S, Saez-Rodriguez J, Gilles ED. Structural and functional analysis of cellular networks with CellNetAnalyzer. BMC Syst Biol, 2007, 1: 2.
[46] Luo R, Liao S, Zeng SQ, et al. Fluxexplorer: a general platform for modeling and analyses of metabolic networks based on stoichiometry. Chin Sci Bull, 2006, 51(6): 689−696.
[47] Paley SM, Karp PD. The pathway tools cellular overview diagram and Omics Viewer. Nucleic Acids Res, 2006, 34(13): 3771−3778.
[48] Cvijovic M, Olivares-Hernández R, Agren R, et al.BioMet toolbox: genome-wide analysis of metabolism. Nucleic Acids Res, 2010, 38(Web Server issue): W144−W149.
Progress in automatic reconstruction and analysis tools of genome-scale metabolic network
Tong Hao1,2,3, Hongwu Ma1,2,3, and Xueming Zhao1,2,3
1 Department of Biochemical Engineering, School of Chemical Engineering & Technology, Tianjin University, Tianjin 300072, China
2 Key Laboratory of Systems Bioengineering, Ministry of Education, Tianjin 300072, China
3 Edinburgh-Tianjin Joint Research Centre for Systems Biology and Synthetic Biology, Tianjin University, Tianjin 300072, China
郝彤, 马红武, 赵学明. 基因组尺度代谢网络自动重构及分析工具研究进展. 生物工程学报, 2012, 28 (6): 661−670.
Hao T, Ma HW, Zhao XM. Progress in automatic reconstruction and analysis tools of genome-scale metabolic network. Chin J Biotech, 2012, 28(6): 661−670.
Received: November 17, 2011; Accepted: March 16, 2012
Supported by: National Basic Research Program of China (973 Program) (Nos. 2012CB725203, 2011CBA00804), National Natural Science Foundation of China (Nos. 21106095, 61100124, 20806055, 20875068), Postdoctoral Science Foundation of China (No. 2011M500512).
Corresponding author: Tong Hao. Tel/Fax: +86-22-27406770; E-mail: haotong@tju.edu.cn
国家重点基础研究发展计划 (973计划) (Nos. 2012CB725203, 2011CBA00804),国家自然科学基金 (Nos. 21106095, 61100124, 20806055,20875068),中国博士后科学基金(No. 2011M500512)资助。
猜你喜欢
Journal of Palaeogeography(2022年1期)2022-03-25
建材发展导向(2022年4期)2022-03-16
快乐语文(2021年35期)2022-01-18
内蒙古民族大学学报(社会科学版)(2020年2期)2020-11-06
铁道通信信号(2018年3期)2018-04-19
摄影之友(影像视觉)(2017年1期)2017-07-18
中国科技信息(2016年10期)2016-09-03
太空探索(2016年5期)2016-07-12
制导与引信(2016年3期)2016-03-20
长江师范学院学报(2015年6期)2015-02-27
