
儿童肝豆状核变性临床表型及ATP7B基因突变关联性分析
2013-11-21 04:50王建设王晓红
中国循证儿科杂志 2013年5期
陆 怡 王建设 俞 蕙 王晓红
肝豆状核变性(WD)系编码铜结合性P型ATP酶的ATP7B缺陷引起的常染色体隐性遗传性疾病。自1993年Bull等发现了WD的基因缺陷以来,国内外学者相继开展了ATP7B基因结构与功能异常的研究。中国汉族人群ATP7B基因研究也有报道,但鲜有儿童方面相关研究被发表。本文对WD患儿进行了ATP7B基因外显子测序分析,以了解其ATP7B基因突变的类型,并探讨临床表型和基因型的相关性。
1 方法
1.1 纳入标准2005年7月至2012年2月复旦大学附属儿科医院确诊WD,且有ATP7B基因检测结果的汉族连续病例。
1.2 WD诊断标准[1]①角膜K-F环,锥体外系症状,血清铜蓝蛋白 <0.1 g·L-1,24h尿铜 >100μg或尿铜正常者青霉胺激发试验尿铜>5倍诊断标准;以上4项指标阳性分别计2分。②Coombs阴性溶血性贫血,24h尿铜40~100μg,肝脏组织铜染色阳性,血清铜蓝蛋白0.1~0.2 g·L-1;以上4项指标阳性分别计1分。③ATP7B基因外显子测序检出纯合突变或复合杂合突变计4分,单一位点杂合突变计1分。总分≥4分,排除其他疾病者为确诊WD病例。
1.3 WD分型 ①根据受累的部位分为:肝病型,有肝病表现,无神经精神症状,分H1和H2亚型;神经型,有神经系统精神系统临床表现或相关影像学检查异常,排除其他神经系统和精神系统疾病,可伴有肝病表现,分N1,N2和Nx亚型。②没有明显的临床表现,因健康体检或其他疾病就诊常规肝功能检查时,发现肝功能损害而进一步检查确诊WD,归为亚临床型;分别因黄疸、水肿、腹胀、关节痛、行走不稳等症状就诊确诊WD为临床型。
1.4 ATP7B基因突变检测 采用PCR技术扩增ATP7B基因全部外显子及邻近序列并直接测序,以NG_008806.1为对照序列,对发现的突变/变异进行反向测序验证。对比dbSNP数据库、1000genome数据库,结合已发表文献和(或)功能预测结果,确定是否为致病性突变。以ENST00000242839为转录模板,确定氨基酸的位置。
1.5 资料截取 从病史中截取以下资料用于分析:①一般情况:性别、就诊年龄、起病年龄等;②临床表型:转氨酶、胆红素、胆汁酸、铜蓝蛋白、24h尿铜水平;角膜K-F环,病理
学检查结果等;③ATP7B基因突变的位置和类型等。
1.6 统计学方法 采用Stata8.0软件进行统计分析,计量资料采用±s表示,组间差异采用t检验;计数资料以百分比表示,率的显著性采用χ2或精确χ2检验。(P<0.05)为差异有统计学意义。
2 结果
2.1 一般情况52个无亲缘关系的家庭中53例WD患儿(其中有一对为姐弟),男34例,女19例,中位起病年龄5.75(2.5~13.5)岁。患儿父母均未确诊WD。死亡1例。
2.2 肝病型和神经型临床表型比较 52/53例(98.1%)有不同程度的肝损害表现,其中肝病型41例,平均起病年龄(6.0±2.3)岁,H1亚型1例(急性溶血性黄疸起病,后出现急性肝功能衰竭,治疗33d后死亡),H2亚型40例。神经型12例,平均起病年龄(9.5±3.0)岁,与肝病型差异有统计学意义(P=0.000 1);N1亚型11例,其中肝脏合并神经系统症状6例(神经系统症状起病就诊者3例),另有5例肝脏损害无明显神经系统症状,但EEG或头颅CT/MRI检查提示有异常表现;Nx型1例,为单纯神经系统受累(肝功能正常,B超肝、脾未发现异常,但未行肝脏穿刺缺乏病理依据)。
表1显示,K-F环阳性16/52例(30.8%),1例(H1亚型)因急性肝功能衰竭无法检测。53例均行铜蓝蛋白、24h尿铜和肝功能检测。肝病型ALT较神经型升高明显;神经型24h尿铜水平、胆汁酸水平和K-F环阳性率均显著高于肝病型((P<0.01))。肝病型2例铜蓝蛋白>2.0g·L-1。31例WD患儿中的60位肝功能正常的父亲和(或)母亲检测铜蓝蛋白水平,其中1例<1.0 g·L-1,20例>2.0 g·L-1。14例(肝病型11例和神经型3例)行肝穿刺,病理均发现纤维化(100%),9例有假小叶形成的肝硬化表现(64.3%),10例(71.4%)脂肪变性,病理误诊为非酒精性脂肪肝2例。
2.3 临床型和亚临床型表型比较 亚临床型34例,均符合肝病型;临床型19例(神经型12例,肝病型7例)。表2显示,亚临床型起病年龄、ALT或AST异常率显著高于临床型,临床型24h尿铜水平、胆汁酸升高比例和K-F环阳性率显著高于亚临床型。
2.4 基因分析 表3显示,53例WD患儿ATP7B基因外显子序列分析发现致病性突变等位基因97个,突变频率为91.5%,8号外显子发现39个等位基因突变(40.2%);13号外显子发现15个等位基因突变(15.5%);11、18号外显子各发现7个等位基因突变(7.2%);5号外显子发现5个等位基因突变(5.2%)。该5个外显子共发现突变等位基因73个,占突变总量的75.3%。
表1 肝病型与神经型的临床表型比较[±s,n(%)]Tab 1 Clinicaldata ofhepatic presentation and neurologic presentation[±s,n(%)]
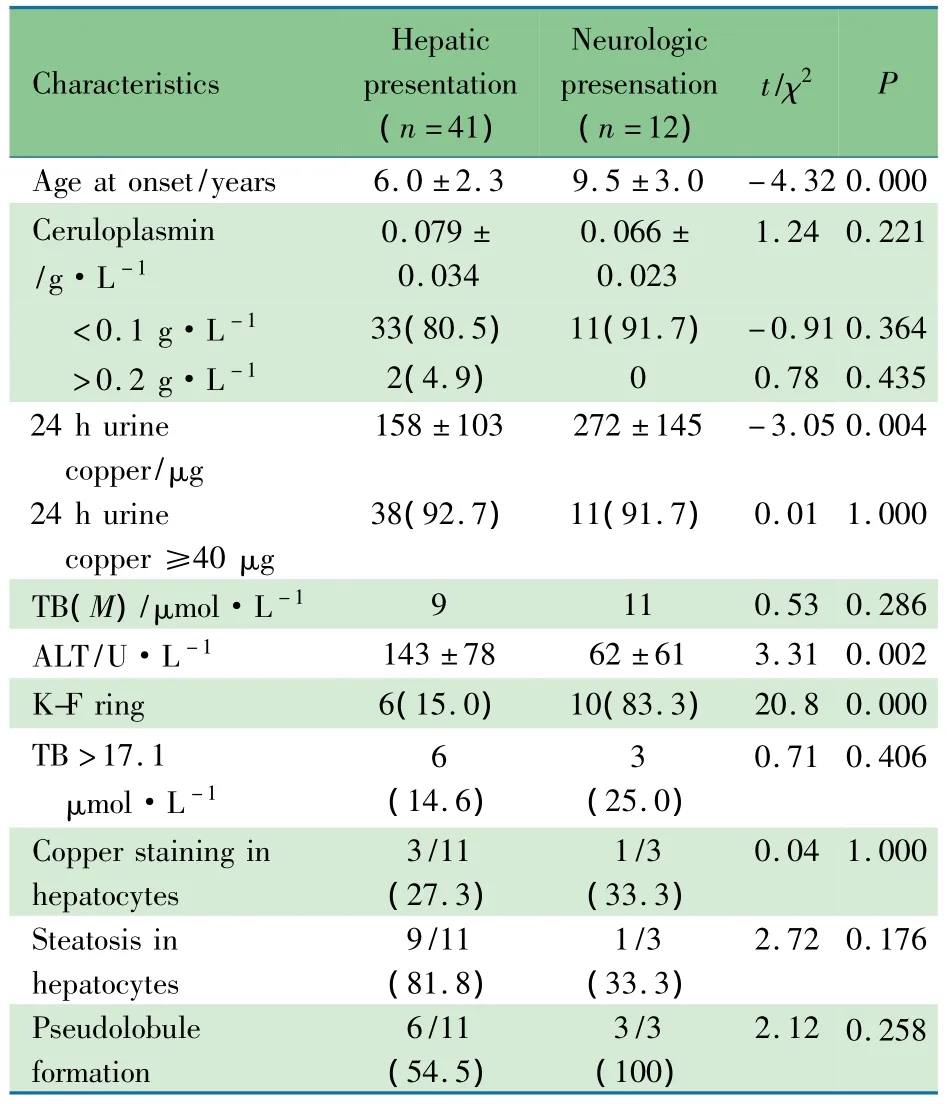
表1 肝病型与神经型的临床表型比较[±s,n(%)]Tab 1 Clinicaldata ofhepatic presentation and neurologic presentation[±s,n(%)]
Characteristics Hepatic presentation(n=41)Neurologic presensation(n=12)t/χ2P Age at onset/years 6.0±2.3 9.5±3.0 -4.320.000 Ceruloplasmin/g·L-1 0.079±0.034 0.066±0.023 1.24 0.221<0.1 g·L-1 33(80.5) 11(91.7) -0.91 0.364>0.2 g·L-1 2(4.9) 0 0.780.435 24h urine copper/μg 158±103 272±145 -3.05 0.004 24h urine copper≥40μg 38(92.7) 11(91.7) 0.011.000TB(M)/μmol·L-1 9 11 0.530.286 ALT/U·L-1 143±78 62±61 3.31 0.002 K-F ring 6(15.0) 10(83.3) 20.80.000TB>17.1μmol·L-1 6(14.6)3(25.0)0.71 0.406 Copper staining inhepatocytes 3/11(27.3)1/3(33.3)0.04 1.000 Steatosis inhepatocytes 9/11(81.8)1/3(33.3)2.72 0.176 Pseudolobule formation 6/11(54.5)3/3(100)2.120.258
表2 临床型与亚临床型的临床表型比较[±s,n(%)]Tab 2 Clinicaldata of clinical type and subclinical type[± s,n(%)]

表2 临床型与亚临床型的临床表型比较[±s,n(%)]Tab 2 Clinicaldata of clinical type and subclinical type[± s,n(%)]
Characteristics Clincal type(n=19)Subclinical type(n=34)t/χ2P Age at onset/years 9.4±3.1 5.2±2.4 5.500.000 Ceruloplasmin/g·L-1 0.071±0.038 0.080±0.057-0.62 0.541 24h urinary copper/μg 276±156 134±79 4.41 0.000 24h urinary copper≥40μg 19(100) 30(88.2) 2.420.284 K-F ring 15/18(83.3) 1(2.9) 35.70.000TB>17.1μmol·L-1 9(47.4)0 19.4 0.000 ALT or AST>40 U·L-1 13(68.4)34(100)12.10.001
53例患儿中纯合突变8例,复合杂合36例,杂合突变9例。8例纯合突变分别为p.Arg778Leu 6例,p.Glu332stop 1例和p.ILe1323Ser 1例(肝衰竭死亡)。检测到36个突变,包括错义突变23种,插入/缺失突变8种,无义突变2种,剪接突变3种。
8号外显子的p.Arg778Leu见于28例的34个等位基因(35.0%,34/97);13号外显子的p.Pro992Leu突变见于15例的15个等位基因(15.5%,15/97);11号外显子的874位点的错义突变(p.Ala874Val、p.Ala874Pro)见于5例的5个等位基因(5.2%,5/97)。
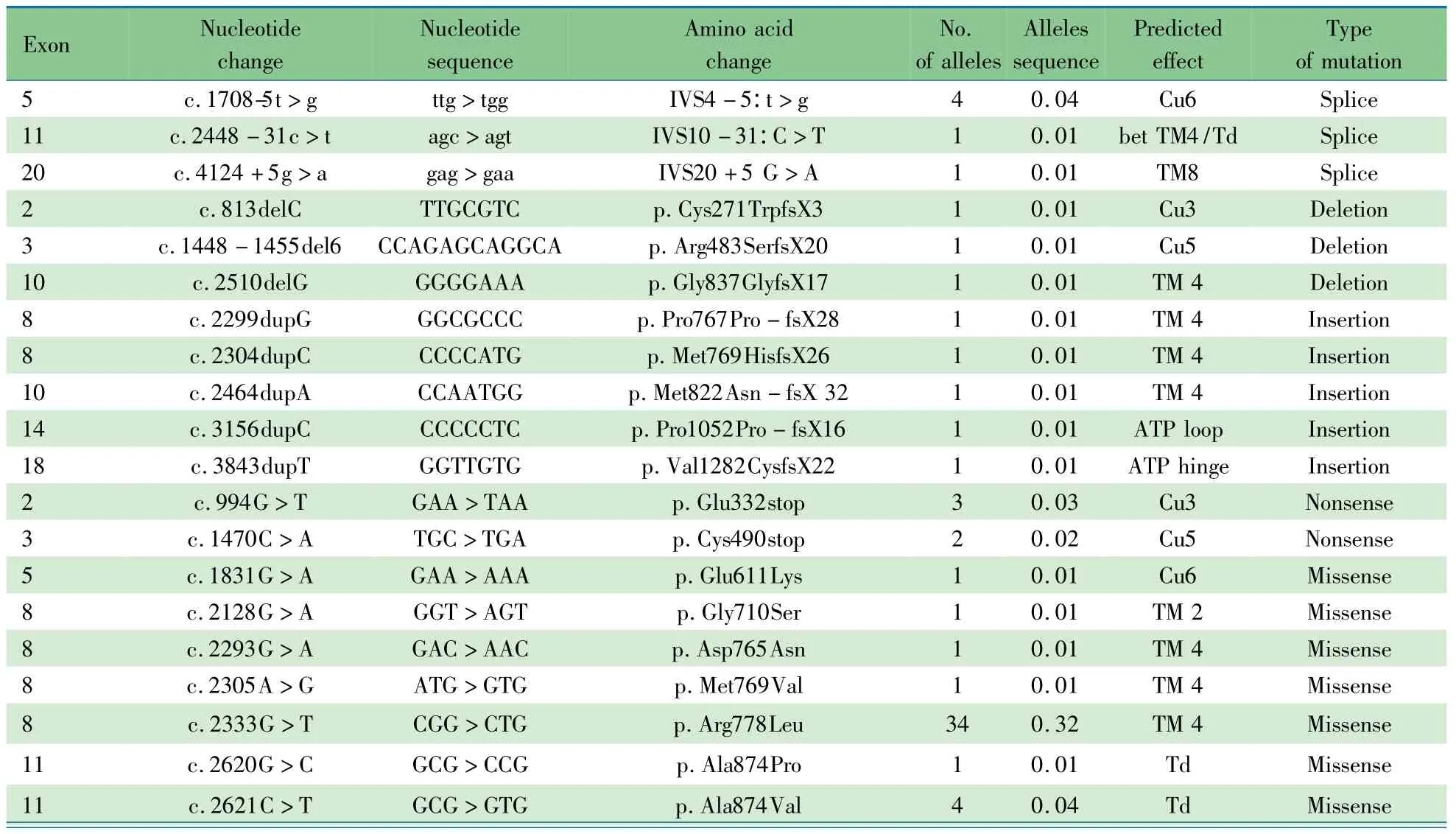
表3 53例WD患儿的ATP7B基因突变Tab 3 ATP7Bgene mutations of 97 pathogenicity alleles in 53 WD cases
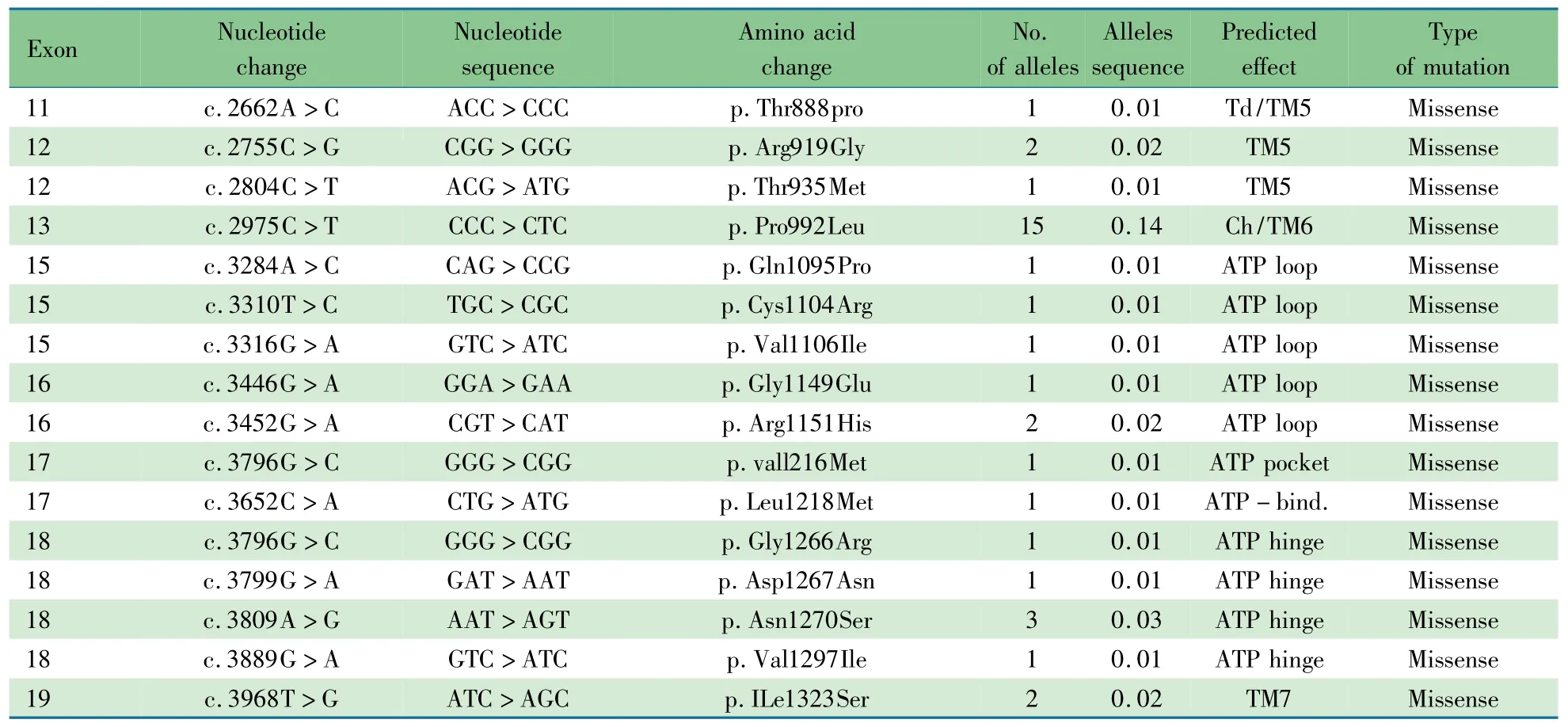
续表3
2.5 基因型和临床表型相关性
2.5.1 突变类型与临床表型的相关性 肝病型41例中纯合突变7例(17.1%),错义突变28例(68.3%),错义+剪接突变33例(80.5%);神经型12例上述3种突变分别为1例(8.3%)、8例(66.7%)和9例(75.0%),肝病型和神经型各突变类型差异无统计学意义,P分别为0.665、1.000和0.697。
临床型19例中纯合突变4例(21.0%),错义突变11例(57.9%),错义+剪接突变13例(68.5%);亚临床型34例中纯合突变4例(11.8%),错义突变24例(70.6%),错义+剪切突变28例(82.4%)。两型不同突变类型差异无统计学意义,P分别为0.301、0.262和0.205。
错义突变与非错义突变,纯合突变与杂合突变的临床表型比较如表4所示,在起病年龄、K-F环阳性率、铜蓝蛋白水平、24h尿铜水平和ALT、AST异常率等方面错义突变与非错义突变,纯合突变与杂合突变差异均无统计学意义(P均>0.05)。纯合突变ALT水平高于杂合突变(P=0.039)
表4 不同突变类型临床表型比较[±s,n(%)]Tab 4 Clinicaldata of patients withdifferent types of mutations[±s,n(%)]
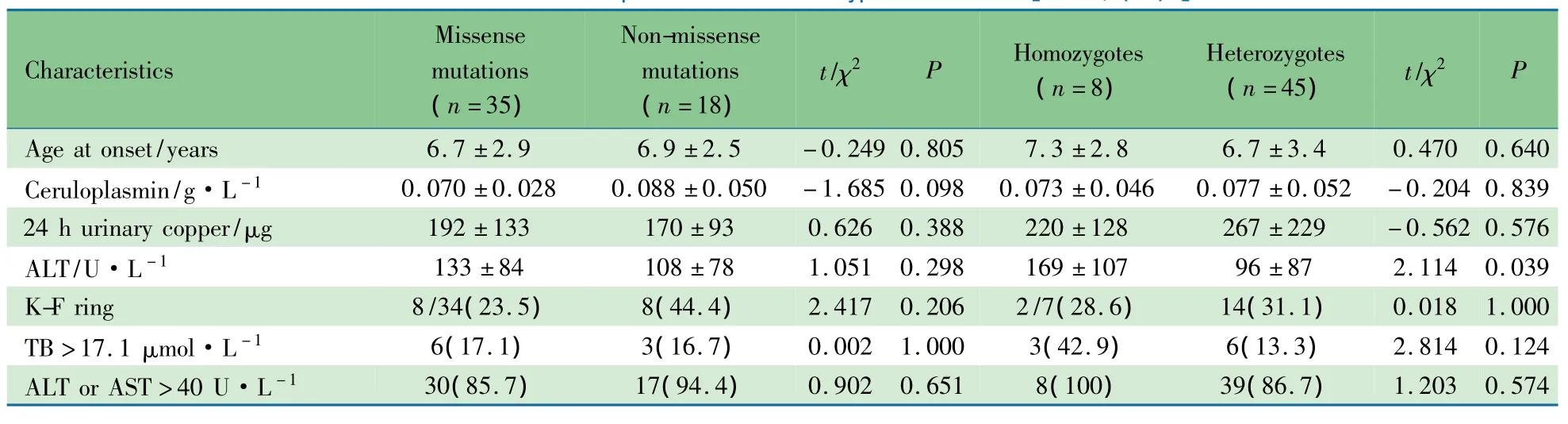
表4 不同突变类型临床表型比较[±s,n(%)]Tab 4 Clinicaldata of patients withdifferent types of mutations[±s,n(%)]
Characteristics Missense mutations(n=35)Non-missense mutations(n=18)t/χ2 P Homozygotes(n=8)Heterozygotes(n=45) t/χ2P Age at onset/years 6.7±2.9 6.9±2.5 -0.249 0.805 7.3±2.8 6.7±3.4 0.4700.640 Ceruloplasmin/g·L-1 0.070±0.028 0.088±0.050 -1.685 0.098 0.073±0.046 0.077±0.052 -0.204 0.839 24h urinary copper/μg 192±133 170±93 0.626 0.388 220±128 267±229 -0.562 0.576 ALT/U·L-1 133±84 108±78 1.051 0.298 169±107 96±87 2.114 0.039 K-F ring 8/34(23.5) 8(44.4) 2.417 0.206 2/7(28.6) 14(31.1) 0.018 1.000TB >17.1μmol·L-1 6(17.1) 3(16.7) 0.002 1.000 3(42.9) 6(13.3) 2.814 0.124 ALT or AST>40 U·L-1 30(85.7) 17(94.4) 0.902 0.651 8(100) 39(86.7) 1.203 0.574
2.5.2 热点突变与临床表型的相关性 肝病型41例中p.Arg778Leu突变26个(31.7%),其中纯合突变5个,杂合突变 16个;神经型 12例中 p.Arg778Leu突变 8个(33.3%),其中纯合突变1个,杂合突变6个,两型差异无统计学意义(P=1.000);p.Pro992 Leu突变在肝病型和神经型分别为13(15.8%)和2个(8.3%),p.Ala874Val/Pro突变肝病型和神经型分别为4(4.9%)和1个(4.2%),均为杂合突变;两型差异均无统计学意义(P分别为0.521和1.000)。
亚临床型34例中p.Arg778Leu突变23个(33.8%),其中纯合突变5个,杂合突变13个;临床型19例中 p.Arg778Leu突变11个(28.9%),其中纯合突变1个,杂合突变9个,两型差异无统计学意义(P=0.668);p.Pro992 Leu突变在亚临床型和临床型分别为11(16.2%)和4个(10.5%),p.Ala874Val/Pro突变在亚临床型和临床型分别为4(5.9%)和1个(2.6%),均为杂合突变;两型差异均无统计学意义(P分别为0.565和0.652)。
p.Ala874Val 4例、p.Ala874Pro 1例,均以肝功能损害起病,平均起病年龄(3.6±0.5)岁;其他肝病型起病年龄(6.3±2.3)岁,神经型起病年龄为(9.5±3.0)岁,差异有统计学意义(P分别为0.017和0.001)。剪接突变6例,其中IVS4-5:t>g突变4例,2例铜蓝蛋白水平 >0.2 g·L-1;IVS10-31:C>T突变1例,铜蓝蛋白水平为0.19 g·L-1;VS20+5 G>A突变1例。p.Glu332stop突变2例,其中1例纯合突变,7.5岁起病,表现为肝功能损害低蛋白血症;1例为与p.Arg778Leu复合杂合突变,10岁以肌张力增高起病,Nx型,2例临床症状均较重。
3 讨论
WD是广泛分布于人类各种族人群中的常染色体隐性遗传性疾病,发病率约3/10万,人群携带率为1/90[21]。Gollan等[3]报道WD的发病率约占活产婴儿的1/30 000,WD患者占人口的15/100万~30/100万,致病基因携带频率为0.3%~0.7%,发病年龄以7~12岁最多见。Mak等[4]计算汉族发病率1/5 400,远高于欧美人群。
WD患者的早期临床症状不一,Gollan等[3]报道首发症状为肝脏损害者占42%,神经精神异常占34%,血液系统症状占12%,肾脏表现占1%,约30%的患者首发症状不易被发现。小年龄者以肝脏症状为主,大年龄者则以锥体外系症状为主。本组资料显示52/53例WD患儿有肝脏损害,以神经系统症状首发起病者仅3例(3/53),肝病型占绝对优势(77.4%),可能和本组小年龄儿童较多有关。
P型ATP7B基因是WD致病基因(ATP7B;OMIM#277900),含21个外显子,cDNA全长约6.6kb,编码1 465个氨基酸。ATP7B有3个主要功能区域[6],第1功能区为铜离子结合区(MBD),位于N端,包括6个与特定铜离子结合的氨基酸保守基序,由第2~5号外显子分别编码,接受从细胞质转运来的铜和帮助把铜释放到跨膜孔道。第2功能区为P型ATP酶功能区,包括磷酸酶活性区与传导区,由第10~11号外显子编码;磷酸化及ATP结合区,由第14~18号外显子编码。第3功能区为8个跨膜区(TMS),间插于整个多肽链中,由第6~8、第12~13及第19~20号外显子分别编码。本文53例共检测到97个突变,涵盖了ATP7B基因的3个功能区,其中以第3功能区的第8号、13号外显子的比例最高,第2功能区的第11号外显子基因突变频率为7.2%,涉及第1功能区的基因突变比例相对较低。
本研究在53例WD患儿中共发现36个突变,包括错义突变23种,插入/缺失突变8种,无义突变2种,剪接突变3种。突变种类繁多,难以对临床表型和每种基因型进行分析。本文通过不同突变类型的临床表现、高发突变位点在不同临床分型的分布,以及不同突变类型与不同临床表型间的对比,试图寻找各种突变临床表型的差异,但均未得到阳性结果。分析原因除了WD突变类型多、形式多样外,还与WD起病隐匿有关,本文亚临床型34例(64.2%),均因健康体检或其他疾病就诊常规肝功能检查时发现肝功能损害,而明确诊断为WD,这部分患儿的早期发现,影响了WD的自然病程,可能对相关性的结果有一定影响;此外部分临床表型也与患儿年龄、治疗和对病情的观察仔细程度等因素有关,也可能会对本文结果产生一定影响。
ATP7B主要有两种丧失转运铜的模式:①ATP7B跨膜转运铜的功能受损;②ATP7B的细胞内异位功能异常。当铜浓度增高时,ATP7B仍位于高尔基体内,但不能将铜转运出细胞。典型变异位点是p.Gly943Ser和p.Met769Val,不影响铜蓝蛋白的形成,这也是少数患者铜蓝蛋白水平不降低的原因[6,7],且中枢神经系统症状轻。ATP7B聚集于细胞边缘,而不是位于正常的TGN内,导致不能结合多余的铜并将其转运出细胞外[8]。最常见导致临床病变的模式是,ATP7B贮留在内质网,不能完成对铜的转运,如最常见的变异位点是 p.His1069Glu 和 p.Arg778Leu[7,8]。
p.Arg778Leu位于ATP7B蛋白质跨膜区(TMS4),与铜运转有关。本研究表明p.Arg778Leu为中国汉族WD患儿最常见的突变。Liu等[9]报道p.Arg778Leu和早期出现肝脏损害症状相关;吴志英等[10]报道p.Arg778Leu纯合突变者起病早、铜蓝蛋白水平较低;提示基因突变和临床表型存在一定的关系。ATP7B基因的778蛋白位密码区是中国人ATP7B基因突变高发区,也是黄种人ATP7B基因突变高发部位。有研究显示,韩国人WD的p.Arg778Leu突变占39.2%[11],日本人为 27%,中国台湾为 28%。Wang等[12]分析了中国南方73例WD患者,p.Arg778Leu是最高发突变(23.3%)。本组R778突变频率占32.1%,与上述研究结果较为一致。但本文进一步分析结果显示,肝病型与神经型p.Arg778Leu突变频率差异无统计学意义,和 Liu等[9]的报道不一致,可能和病例来源的不同有关。
p.Pro992Leu是本文病例第2位常见致病突变,占14.2%。位于13号外显子的蛋白质跨膜区(TMS6),与铜转运相关;该位点是黄种人主要致病基因。但Wang等[12]的报道I1148T的突变频率9.6%(占第2位),而本研究未发现1148位点的突变;这种差异是否为样本组成不同所致,仍有待进一步分析。
p.Ala874Val/Pro是本研究第3位的常见基因突变,其中该位点的p.Ala874Val已多有报道,p.Ala874Pro相对少见。Park等[13]报道韩国 WD病例的常见突变为 p.Arg778Leu,p.Ala874Val和 p.Asn1270Ser,提示874 位点的突变在亚洲人群中并不罕见,与本文相符。本文因p.Ala874Val/Pro例数较少,未行基因频率的统计学分析,但发现p.Ala874Val/Pro发病年龄显著低于其他肝病型以及神经型患儿,查阅其对应功能区为Td,为ATP7B基因的磷酸酶活性与传导区域,靠近第5号跨膜蛋白(TM5),该区位于两个跨膜区之间(TM4~5),该区的突变可导致蛋白高度磷酸化。有研究显示,ATP7B催化过程中蛋白跨膜部分和胞浆部分构象改变时,A区(包含Td)的活动显著变化,据此猜测MBD5~6结合铜离子后,影响了A区的活动,从而触发跨膜部分构象改变,影响蛋白反转继而阻断铜离子的跨膜运转。
刘晓青等[14]报道2例 p.Val1106Ile突变 WD患者均为晚发型,1例为 p.Arg778Leu/Val1106Ile,1例为 p.Val1140Ala/Val1106Ile。本组病例检出1例p.Arg778Leu/p.Val1106Ile,起病年龄仅3岁,临床因其他原因就医时意外发现肝脏问题而诊断WD,与文献报道病例的临床表现不一致。本组还发现p.Glu332stop 2例学龄期起病者,临床症状均比较重,有待进一步研究。本研究发现剪接突变的6例WD患儿的铜蓝蛋白值明显高于其他突变类型,尤其IVS4-5:t>g突变。提示剪接突变影响了ATP7B基因在细胞内的异位功能,而对铜离子的跨膜转运影响小。
随着对ATP7B基因研究的深入,不同部位的功能也逐步明了,理论上不同的突变类型会有不同类型和程度的功能受损,临床上会有差异体现。但已有文献报道WD同卵双生者,临床不同的表现形式。推测WD临床表型除受基因影响外还与环境因素等相关,这也增加了基因型与表型关系建立的难度。
结论:本研究显示,WD患儿几乎均有肝脏受累,多以肝病表现起病。ATP7B基因常见突变为p.Arg778Leu、p.Pro992Leu和p.Ala874Val,常见基因型和临床表型间未发现显著相关性。874蛋白位点的变异者起病年龄较早,剪接突变对铜蓝蛋白的影响可能较小。
致谢本研究基因分析得到上海交通大学医学院附属瑞金医院张欣欣教授、李新华博士的大力支持,在此特别致谢!
[1]Ferenci P, Caca K, Loudianos G, et al.Diagnosis and phenotypic classification of Wilsondisease.Liver Int, 2003,23(3):139-142
[2]Figus A,Angius A,Loudianos G,et al.Molecular pathology andhaplotype analysis of Wilsondisease in Mediterranean populations.Am J Hum Genet, 1995,57(6):1318-1324
[3]Gollan JL, GollanTJ.Wilson disease in 1998:genetic,diagnostic and therapeutic aspects.J Hepatol, 1998,28(S1):28-36
[4]Mak CM, Lam CW, Tam S, et al. Mutational analysis of 65 Wilson disease patients in Hong Kong Chinese: identification of 17 novel mutations and its genetic heterogeneity. J Hum Genet,2008,53( 1) : 55-63
[5]Chen DB(陈定邦), Feng L, Li XH.Wilson'sdisease:recent advancement in molecular biology.Journal of Molecular Diagnosis andTherapy(分子诊断与治疗杂志),2011,3(2):120-124
[6]Lalioti V, Sandoval I, Cassio D, et al.Molecular pathology of Wilson's disease:a brief.J Hepatol, 2010,53(6):1151-1153
[7]Forbes JR, Cox DW.Copper-dependent trafficking of Wilsondisease mutant ATP7B proteins.Hum Mol Genet, 2000,9(13):1927-1935
[8]Huster D, Hoppert M, Lutsenko S, et al. Defective cellular localization of mutant ATP7B in Wilson's disease patients and hepatoma cell lines. Gastroenterology, 2003,124( 2) : 335-345
[9]Liu XQ, Zhang YF, Liu TT, et al. Correlation of ATP7B genotype with phenotype in Chinese patients with Wilson disease. World J Gastroenterol,2004,10( 4) : 590-593
[10]Wu ZY(吴志英),Wang N, Lin MT,et al.Genotype phenotype correlation of patients with wilsondisease in Chinese population.Natl Med J China(中华医学杂志),2003,83(4):309-311
[11]Park S, Park JY, Kim GH, et al.Identification of novel ATP7B gene mutations and their functional roles in Korean patients with Wilson disease.Hum Mutat, 2007,28(11):1108-1113
[12]Wang LH, Huang YQ,Shang X, et al.Mutation analysis of 73 southern Chinese Wilson's disease patients:identification of 10 novel mutations and its clinical correlation.J Hum Genet,2011,56(9):660-665
[13]Park HD, Ki CS, Lee SY, et al.Carrier frequency of the R778L,A874V,and N1270S mutations in the ATP7B gene in a Korean population.Clin Genet, 2009,75(4):405-457
[14]Liu XQ(刘晓青),Zhang YF,Liu ZZ, et al.Genotype and phenotype correlation in Chinese patients with Wilson's Disease.Chin J Pediatr(中华儿科杂志),2003,41(1):35-38
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
广西医科大学学报(2022年5期)2022-06-07
昆明医科大学学报(2022年3期)2022-04-19
种子(2021年3期)2021-04-12
中国生殖健康(2020年4期)2021-01-18
中南医学科学杂志(2019年6期)2019-12-05
中国生殖健康(2018年4期)2018-11-06
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
中学生理科应试(2016年7期)2016-05-14
