
米根霉AS 3.819基因启动子片段的克隆及功能鉴定
2015-03-17 05:36姜绍通潘丽军李兴江罗水忠吴学凤
食品科学 2015年7期
张 旻,姜绍通*,郑 志,潘丽军,李兴江,罗水忠,吴学凤
(合肥工业大学生物与食品工程学院,安徽省农产品精深加工重点实验室,安徽 合肥 230009)
米根霉AS 3.819基因启动子片段的克隆及功能鉴定
张 旻,姜绍通*,郑 志,潘丽军,李兴江,罗水忠,吴学凤
(合肥工业大学生物与食品工程学院,安徽省农产品精深加工重点实验室,安徽 合肥 230009)
摘 要:从L-乳酸生产菌米根霉(Rhizopus oryzae)菌株AS 3.819基因组DNA中分别扩增得到了乳酸脱氢酶基因(ldhA)、丙酮酸脱氢酶基因(pdcA)、淀粉糖化酶基因(amyA)以及磷酸甘油酸酯激酶基因(pgk1)的启动子片段,并构建启动子探针载体pUKMR,以β-内酰胺酶基因(bla)为报告基因在大肠杆菌JM109中对这些启动子片段进行筛选及启动活性检测。结果表明:4 种启动子片段成功启动报告基因表达;在非底物诱导情况下,ldhA和pgk1启动子启动活性较强;在有合适碳源底物诱导情况下pdcA和amyA启动子拥有更高的启动活性;ldhA基因的启动子启动活性随着启动子片段长度的增加有一定提高,而在长度为500 bp以上时,其启动活性变化不明显。本研究为Rhizopus oryzae提供了一种快速简便的启动子捕获分离及启动活性检测方法。
关键词:米根霉;大肠杆菌;启动子探针载体;β-内酰胺酶;乳酸脱氢酶
米根霉(Rhizopus oryzae)是一种重要的工业微生物,能够利用葡萄糖等己糖、木糖等戊糖、淀粉及木质纤维素等碳源生产高纯度L-乳酸[1-6]及其他有机酸[7-8],在食品、医药、化学工业、皮革、香料等领域具有广泛的用途。同其他许多丝状真菌一样,由于培养条件简单,具有高效的分泌表达系统以及易于实现大规模连续发酵等优点,米根霉也有着应用于同源或者异源蛋白表达,成为一种高效基因工程菌的潜力。但是由于稳定可靠的针对野生型或者工业米根霉菌株的遗传转化方法尚未发展成熟,对米根霉的遗传改造工作目前仍然存在较大的难度。
启动子是基因表达调控的重要元件,对于基因表达水平有着重要的影响。随着丝状真菌遗传改造研究越来越深入,越来越多的真菌基因的启动子被分离及鉴定,并被应用于真菌表达质粒的构建当中。对于米根霉而言,获取高效稳定或者可调控的启动子对于米根霉表达载体构建也是十分重要的。启动子片段的分离一般采用启动子探针质粒捕获的方法,包括从基因组文库筛选以及从聚合酶链式反应(polymerase chain reaction,PCR)扩增产物中筛选启动子片段,而启动子片段的克隆方法又包括常规PCR法、反向PCR法、锅柄PCR法、序列特异性引物PCR法、热不对称交错PCR法和Y型接头扩增法等[9]。启动子探针质粒依靠其中的报告基因对启动子片段进行筛选以及功能分析[10-13]。在启动子探针质粒构建当中,常常选择绿色荧光蛋白(green fluorescent protein)基因[14]、β-半乳糖苷酶(β-galactosidase)基因[15]及β-葡萄糖醛酸苷酶(β-glucoronidase)基因[16]作为报告基因。另外一种类型的启动子探针载体则是以抗性标记基因作为报告基因,包括氯霉素抗性基因、卡那霉素抗性基因、氨苄青霉素抗性基因以及潮霉素抗性基因等[17-20]。
本研究以pUC18克隆载体为载体构建基础,β-内酰胺酶(β-lactamase)基因bla(氨苄青霉素抗性基因)为报告基因构建探针质粒。应用此探针载体在大肠杆菌JM109当中进行米根霉基因启动子的筛选,并通过测定转化子氨苄青霉素耐受性,β-内酰胺酶活性,检测并比较几种启动子片段的启动活性,以期为米根霉或其他丝状真菌的启动子片段快速简便的捕获分离方法提供参考。
1 材料与方法
1.1菌株
米根霉(Rhizopus oryzae)AS 3.819菌株 中国科学院微生物研究所(菌种保藏编号:CICC40313);大肠杆菌Escherichia coli JM109 宝生物工程(大连)有限公司。
1.2质粒
本研究所用到的质粒均列于表1中。

表1 本研究所用质粒Table 1 Plasmids used in this study
1.3试剂与仪器
1.3.1 试剂
rTaq DNA聚合酶,DNA限制性内切酶SspⅠ、NdeⅠ、EcoRⅤ和SmaⅠ,T4连接酶,DNA Marker,异丙基-β-D-硫代半乳糖苷(isopropyl β-D-thiogalactoside, IPTG)、5-溴-4-氯-3-吲哚-β-D-半乳糖苷(5-bromo-4-chloro-3-indolyl β-D-galactoside,X-Gal)、去磷酸化酶(alkaline phosphatase)来自于E. coli C75以及pUC18载体 日本TaKaRa公司;硫酸卡那霉素(kanamycin)北京Solarbio公司;氨苄青霉素(ampicillin) 美国Amersco公司;β-内酰胺酶标准品 美国Sigma公司;青霉素钾盐 中国药品生物制品检定所;PCR产物胶回收纯化试剂盒 美国Axygen公司;质粒小量抽提试剂盒以及pEASY-T1克隆载体试剂盒 北京全式金生物工程有限公司;Amicon Ultra-15超滤管及超滤膜 美国Merck Millipore公司。
1.3.2 仪器与设备
TC-96 PCR仪 杭州博日科技有限公司;Universal HoodⅡ凝胶成像系统 美国Bio-Rad公司;Himac CR22GⅡ高速冷冻离心机 日本Hitachi公司;TU-1901紫外分光光度计 北京普析分析仪器公司。
1.4引物
根据米根霉基因组序列数据库(http://www. broadinstitute.org/annotation/genome/rhizopus_ oryzae/MultiHome.html)中公布的各个基因的旁侧序列,设计引物ldhpF以及ldhpR(两端引入SmaⅠ酶切位点)扩增ldhA基因的启动子区域片段PldhA;设计引物pdcpF以及pdcpR(两端引入SmaⅠ酶切位点)扩增pdcA基因的启动子区域片段PpdcA;设计引物amypF及amypR(两端引入EcoRⅤ酶切位点)扩增amyA基因的启动子区域片段PamyA;设计引物pgkpF以及pgkpR(两端引入SmaⅠ酶切位点)扩增pgk1基因的启动子区域片段Ppgk1;根据pEASY-T1质粒的序列设计引物KmrU(引入SspⅠ及EcoRⅤ酶切位点)及KmrD(引入NdeⅠ酶切位点)从pEASY-T1质粒上扩增卡那霉素抗性基因及其上下游序列。以上引物均由生工生物工程(上海)股份有限公司进行合成。引物序列列于表2中。

表2 PCR所用引物及其序列Taabbllee 2 Primers used for PCR process and their sequences
1.5方法
1.5.1 米根霉基因启动子片段分离
从米根霉斜面上制取孢子乳悬液,并按体积分数5%的接种量接种到培养基中。米根霉培养所用培养基成分如下:葡萄糖120 g/L、硫酸铵4 g/L、磷酸二氢钾0.3 g/L、磷酸二氢钠0.3 g/L、硫酸锌0.44 g/L、硫酸镁0.25 g/L。32 ℃振荡培养12 h左右。当培养液中有均匀分散的米根霉菌丝体时便可取出进行基因组DNA的提取。
米根霉菌体基因组提取过程参照分子克隆实验指南有关内容并加以改进[21]。以基因组DNA为模板进行各个启动子片段的扩增。扩增片段的回收纯化、连接、转化大肠杆菌、质粒提取、酶切及回收等过程参考分子克隆实验指南。
1.5.2 启动子探针载体的构建
启动子探针质粒的骨架来源于pUC18克隆载体见表1。用限制性内切酶SspⅠ以及NdeⅠ酶切pUC18质粒,切除368 bp含有β-内酰胺酶基因的启动子的片段,使此基因无法表达,并获得含有SspⅠ及NdeⅠ酶切黏性末端的线性质粒。同时将用引物KmrU及KmrD扩增并克隆得到的1.1 kb带有卡那霉素抗性基因(kan)的表达盒片段与其进行连接(此片段5’端含SspⅠ及EcoRⅤ酶切位点,3’端含NdeⅠ酶切位点),转化大肠杆菌并用40 µg/mL的卡那霉素进行筛选,由此得到探针质粒pUKMR。此质粒的原SspⅠ酶切位点相邻处存在由引物引入的EcoRⅤ位点,可通过此酶切位点插入待筛选的平末端启动子片段,并可能启动β-内酰胺酶基因的表达,使宿主菌获得氨苄青霉素抗性。
1.5.3 米根霉基因启动子在大肠杆菌E. coli JM109中的启动活性检测
1.5.3.1 启动子片段的筛选
构建的启动子探针质粒经过酶切、去磷酸化处理之后回收,与同样经过酶切回收的启动子片段进行过夜连接并转化大肠杆菌E. coli JM109,以氨苄青霉素为抗性标记筛选得到含重组质粒的抗性转化子。挑选抗性转化子菌落于含50 µg/mL的氨苄青霉素的液体LB培养基中培养并提取质粒,并通过PCR检测及测序检测确认是否连入了启动子片段。
筛选并鉴定出的转化子经培养之后采用平板划线的方式接种于含不同质量浓度(50、80、100、150、180、200、220、250、300 µg/mL)的氨苄青霉素的固体LB平板上,观察β-内酰胺酶基因在各个启动子片段启动作用下的表达导致转化子对氨苄青霉素的耐受性,并由此初步考察这几种启动子片段的启动活性。
1.5.3.2 β-内酰胺酶活力检测
β-内酰胺酶活力检测参考文献[22]方法,取对氨苄青霉素具有抗性的上述菌落,接种在含有40 µg/mL的卡那霉素的50 mL的LB液体培养基中,于37 ℃、180 r/min摇瓶培养18 h达到对数生长后期收集菌液。8 000 r/min离心10 min,收集培养上清液,通过Amicon Ultra-15超滤管(超滤膜截留分子质量10 000 D)超滤浓缩10 倍至5 mL。浓缩后的上清用以测定β-内酰胺酶活力。酶活力测定采用以青霉素钾为底物,2,2’-联喹啉为显色剂的方法,在分光光度计下检测反应后颜色对比计算样品中β-内酰胺酶活力。测定方法如下:1 mL含5 000 U/mL青霉素钾的醋酸盐缓冲液(20 mmol/L,pH 6.0)中加入0.1 mL浓缩上清样品,37 ℃反应1 h。反应结束后添加1 mL的13 g/100 mL三氯乙酸溶液,混合均匀后于4 000 r/min离心10 min,取上清0.2 mL,添加0.05 g/100 mL的2,2’-联喹啉4.8 mL,混合均匀后添加20 mmol/L的CuSO4溶液0.4 mL,混合均匀室温静置显色15 min后,在分光光度计中于550 nm波长处测定吸光度,空白对照中样品替换为等量LB液体培养基。并以β-内酰胺酶标准品制作酶活力-质量浓度标准曲线,计算浓缩样品酶活力(U/mL)。1.5.3.3 蛋白质含量测定
测定1.5.3.2节中浓缩处理得到的培养上清液中的可溶蛋白质含量,测定方法参照Bradford法[23]。
2 结果与分析
2.1基因启动子片段克隆

图1 米根霉AS 3.819基因启动子片段PCR扩增产物Fig.1 PCR amplification products of gene promoter fragments from R. oryzae AS 3.819
以米根霉AS 3.819基因组DNA为模板,ldhpF及ldhpR为引物扩增PldhA片段,得到约250 bp大小的特异性条带(图1A);pdcpF及pdcpR为引物扩增PpdcA片段,获得约1 000 bp大小的特异性条带(图1B);amypF及amypR为引物扩增PamyA片段,获得约900 bp大小的特异性条带(图1C);pgkpF及pgkpR为引物扩增Ppgk1片段,获得约500 bp大小的特异性条带(图1D)。从电泳结果来看,与设计中4 条片段的260、1 080、896、541 bp长度相符合。4 条片段送往生工生物工程(上海)股份有限公司进行测序,测序结果对比NCBI以及米根霉基因组序列数据库相应片段的序列,结果表明扩增片段序列正确。
2.2启动子探针质粒的构建与鉴定
如1.5.2节部分所述,本研究中所构建的探针载体pUKMR在其β-内酰胺酶编码基因的上游缺失启动子部分,此载体无法使宿主获得氨苄青霉素抗性。只有在此位点插入具有启动功能的片段才能重新让β-内酰胺酶编码基因在其调控之下进行表达,并通过氨苄青霉素抗性筛选出转化子。
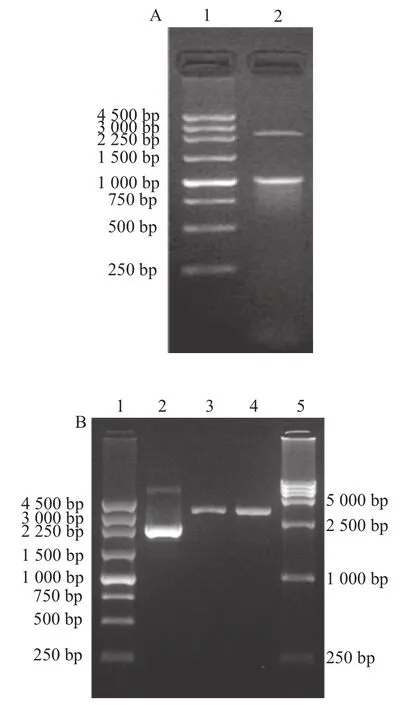
A图:1. 250 bp DNA Ladder Marker标准,2. 含kan基因的表达盒片段的PCR产物;B图:1. 250 bp DNA Ladder Marker标准,2. pUKMR质粒提取,3、4. pUKMR质粒EcoR Ⅴ单酶切后产物,5. DL15000 DNA Marker标准。
图 22 kkaann基因表达盒PCR扩增与质粒pUUKKMMRR的EEccooRⅤ单酶切鉴定结果
Fig.2 PCR amplification of kan gene and restriction enzyme digestion
test of plasmid pUKMR with EcoRⅤ
卡那霉素抗性基因(kan)是以pEASY-T1质粒为模板扩增获得的(图2A),替换pUC18载体上的β-内酰胺酶编码基因(bla)的启动子部分构建成pUKMR质粒。通过卡那霉素抗性筛选出转化子,将转化子分别接种于含40 µg/mL卡那霉素及含50 µg/mL氨苄青霉素的LB液体培养基中进行比较,结果转化子能在含40 µg/mL卡那霉素的LB液体培养基中生长,而无法在含50 µg/mL氨苄青霉素的LB液体培养基中生长,说明筛选出的转化子确实已经不具备氨苄青霉素抗性了,可以认为探针载体pUKMR的bla上游是缺失启动子的。质粒经过EcoRⅤ限制性内切酶单酶切并回收后将产生的平末端进行去磷酸化处理,获得带平末端的探针载体,便可与同样平末端的启动子片段进行连接。从经过EcoRⅤ限制性内切酶单酶切后产生的线性载体的电泳结果来看,大小与预期的3 349 bp大小相符合(图2B)。
2.3各启动子片段在大肠杆菌E. coli JM109中的启动功能
2.3.1 启动子的筛选及其启动活性比较
由于在PldhA、PpdcA、PamyA及Ppgk1片段的两端引入了EcoRⅤ或SmaⅠ位点,因此通过EcoRⅤ或SmaⅠ酶切获得带平末端的启动子片段。通过T4连接酶将其与去磷酸化的线性平末端探针载体pUKMR过夜连接,之后转化大肠杆菌E. coli JM109,并且通过50 μg/mL的氨苄青霉素进行筛选,获得了pUK-PldhA、pUK-PpdcA、pUK-PamyA及pUK-Ppgk1转化子。转化子质粒分别经过了PCR和酶切验证,表明启动子片段都已插入到了探针载体pUKMR上并替换了β-内酰胺酶基因原来的启动子。
将成功转化pUK-PldhA、pUK-PpdcA、pUK-PamyA及pUK-Ppgk1重组质粒而获得氨苄青霉素抗性的转化子分别采用平板划线的方式接种于含有50、80、100、150、180、200、220、250、300 µg/mL氨苄青霉素的固体LB平板上于37 ℃培养12 h,经过抗性检测比较3 种转化子对氨苄青霉素的耐受能力,并以转化pUC18质粒的E. coli JM109为对照。由表3可知,转化入pUK-PldhA的转化子可以耐受最高达200 µg/mL的氨苄青霉素,而使用pUK-PpdcA以及pUK-PamyA得到的转化子其耐受氨苄青霉素能力均比pUC18转化子低,在150 µg/mL 氨苄青霉素的LB平板中已经无法生长,这4 种转化子中以pUK-Ppgk1转化子耐受性最高,能耐受高达250 µg/mL的氨苄青霉素。

表3 含不同启动子的转化子对氨苄青霉素的耐受性Table 3 Ampicillin resistance of recombinants containing different promoters
2.3.2 含启动子片段的转化子β-内酰胺酶活性比较

图3 不同启动子作用下表达的β-内酰胺酶活性Fig.3 Activity of β-lactamase expressed under the control of different promoters
筛选出的含4 种启动子的重组转化子,分别经过含40 μg/mL卡那霉素的LB培养基培养后,按照1.5.3.2节方法测定转化子分泌的β-内酰胺酶活力。如图3所示,含不同启动子的转化子其胞外蛋白中β-内酰胺酶的活力及比活力均有所差别,其表达的β-内酰胺酶量有所差别,间接可以反映出各个启动子启动β-内酰胺酶基因表达强度的不同。其中以携带pgk1基因启动子的转化子其表达的β-内酰胺酶活性最高,其次为含ldhA启动子的转化子。而amyA及pdcA基因启动子在此条件下启动能力低于pUC18中原β-内酰胺酶基因bla自带的启动子。此结果与2.3.1节中各个转化子对氨苄青霉素的耐受性比较结果相吻合。通过此结果可以得知,在LB培养基的培养条件下,启动子启动能力为Ppgk1>PldhA>PpdcA>PamyA,其中PamyA和PpdcA启动能力低于pUC18中自带的启动子。
2.4启动子在不同碳源下的启动活性
微生物代谢过程中,许多碳代谢相关途径中的关键酶的表达往往会受到培养底物的诱导或抑制,其原因之一可能是由于代谢底物自身或者代谢中间物对于相关酶基因启动子区域上的元件的作用使得基因的表达得到增强或抑制,这也是微生物发酵代谢调控的重要基础之一。基因表达类型分为诱导型表达、组成型表达和组织特异型表达,主要取决于启动子启动基因表达的模式。其中诱导型表达的基因其启动子在培养过程中未添加诱导物的情况下,不启动基因表达或者以一定的水平进行本底表达。这在代谢调控研究中有着十分重要的意义,可是使微生物在不同生长条件下合理分配代谢流量在代谢网络中的分布,节省自身能量消耗并且充分利用资源。
在包括米根霉在内的丝状真菌中,碳代谢网络相关的部分酶蛋白基因的表达类型属于诱导型表达,其表达量随底物中碳源的不同存在很大的区别。从2.3.2节结果得知,在LB培养基培养条件下amyA和pdcA基因启动子启动bla表达的能力较低。本研究比较了在不同碳源作为培养底物情况下各启动子的启动活性的变化。在含不同质量浓度梯度氨苄青霉素的LB筛选培养基中各添加了葡萄糖、蔗糖、麦芽糖、甘油及淀粉至终质量浓度为5 g/L,分别在这些平板上划线接种含不同启动子的宿主菌株,观察生长情况。如表4~7所示,PpdcA和PamyA在不同碳源底物诱导作用下表现出一定的启动活性差别,使得宿主大肠杆菌氨苄青霉素耐受性出现变化。其中PpdcA启动活性在葡萄糖和蔗糖中得到了一定的提升,在淀粉和麦芽糖中启动活性与普通筛选LB相比无显著变化,而在甘油中其启动活性丧失,宿主菌无法在含有甘油的LB筛选平板上生长。PamyA则在淀粉中启动活性表现出较高的增强,在葡萄糖及蔗糖中启动活性有一定的增加,在甘油及麦芽糖中启动活性与普通LB筛选培养基中相比基本无差别。相比之下,PldhA和Ppgk1的启动活性则受碳源底物种类的影响则不大,除甘油中Ppgk1启动活性受到较大影响外,其他筛选培养基下生长状况和普通LB筛选培养基中无明显区别。

表4 不同碳源底物下含llddhhAA基因启动子的转化子对氨苄青霉素的耐受性Table 4 Ampicillin resistance of recombinants containing ldhA promoter with different carbon substratess

表5 不同碳源底物下含pdcA基因启动子的转化子对氨苄青霉素的耐受性Table 5 Ampicillin resistance of recombinants containing pdcA promoter with different carbon substratess
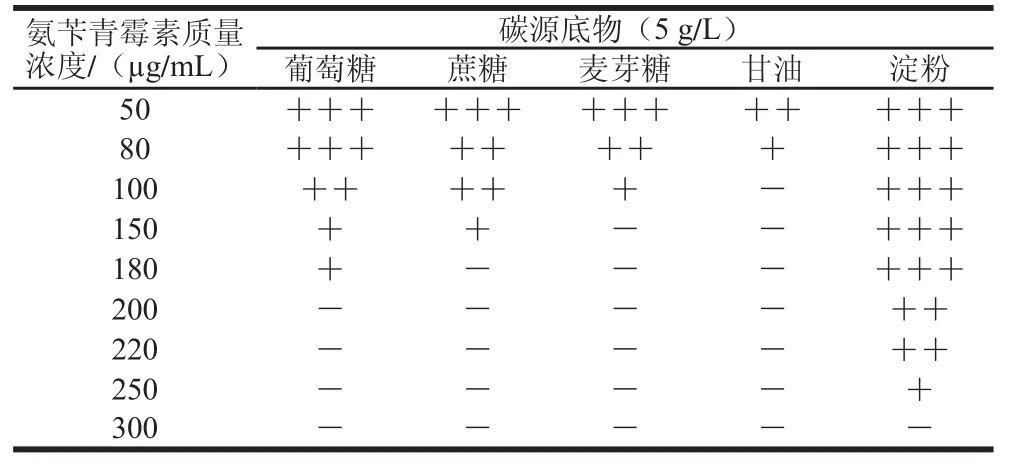
表6 不同碳源底物下含aammyyAA基因启动子的转化子对氨苄青霉素的耐受性TTaabbllee 66 Ampicillin resistance of recombinants containing aammyyAA promoter with different carbon substratess

表7 不同碳源底物下含ppggkk11基因启动子的转化子对氨苄青霉素的耐受性TTaabbllee 7 Ampicillin resistance of recombinants containing ppggkk11 promoter with different carbon substrates
此外,依照1.5.3.2节的方法,在各添加了葡萄糖、蔗糖、麦芽糖、甘油及淀粉至终质量浓度为5 g/L的含40 μg/mL卡那霉素的液体LB培养基中培养宿主菌,分析其表达的β-内酰胺酶活性。如图4所示,含PamyA的宿主菌在淀粉及葡萄糖中其表达的β-内酰胺酶活性确实得到了很大的提高,同样含PpdcA的宿主菌在葡萄糖和蔗糖中β-内酰胺酶活性也有提升,而含PldhA和Ppgk1的宿主菌除在甘油条件下以外,随着碳源底物改变其启动表达的β-内酰胺酶活性变化不大。因此对比2.3节部分所得出的结果,可以认为米根霉的amyA及pdcA基因属于诱导型表达,其基因启动子的启动活性受到碳源底物的诱导及抑制作用的调控,其在各自强诱导底物的作用下启动活性达到相对较高水平。

图4 不同碳源底物下4 种启动子启动表达的β-内酰胺酶活性Fig.4 Activity of β-lactamase expressed under the control of four promoters with different carbon substrates
2.5ldhA基因启动子片段长度对于启动活性的影响
本实验研究了未曾应用于真菌表达载体构建的乳酸脱氢酶A(ldhA)基因启动子,对其启动活性进行了检测。同时也发现这种启动子启动活性相较其他的启动子(包括受到诱导情况下的PamyA和PpdcA)并不高。除了在本研究所设置的诱导条件下启动活性未表现出受诱导的现象之外,其相对较短的长度也有可能会致使其启动能力未得到完全的发挥。因此本研究对ldhA启动子片段长度进行了增加,搜寻了ldhA上游序列中合适的扩增位点,分别使用上游引物ldhpF2、ldhpF3、ldhpF4、ldhpF5以及下游引物ldhpR扩增了长度分别为529、777、1 104、1 363 bp的ldhA启动子片段(分别为PldhA2、PldhA3、 PldhA4、PldhA5),并连入了启动子探针质粒pUKMR中。通过50 μg/mL的氨苄青霉素筛选出抗性菌落,并经过40 μg/mL的卡那霉素和50 μg/mL的氨苄青霉素验证以及PCR验证后得到连入启动子的转化子。
在非诱导条件下对含有不同长度ldhA启动子片段的宿主的氨苄青霉素耐受性和表达β-内酰胺酶活性分别进行比较。如表8和图5所示,当ldhA启动子片段长度由260 bp增加到529 bp时,启动子片段的启动活性有些许增强,而继续增加启动子其启动活性却不再增加,整体看来ldhA启动子长度的增加对于bla基因在大肠杆菌中表达的增强并不十分明显。
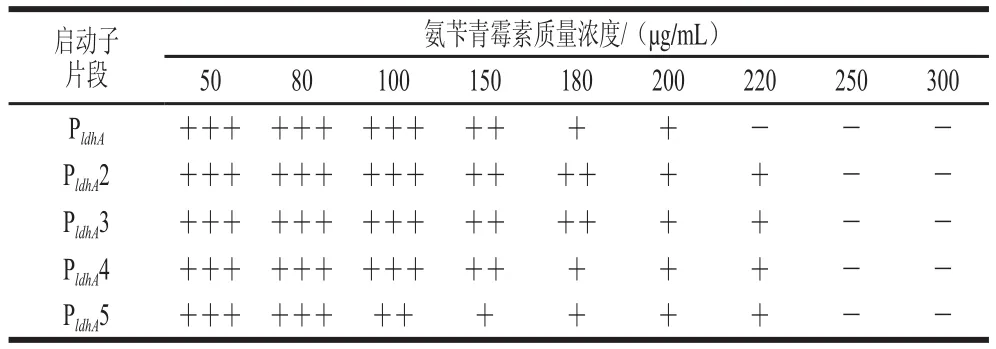
表8 含不同长度的llddhhAA启动子片段的转化子对氨苄青霉素的耐受性Taabbllee 8 Ampicillin resistance of recombinants containing llddhhAA promoter fragments of different lengths

图5 不同长度的llddhhAA启动子片段作用下表达β-内酰胺酶活性Fig.5 Activity of β-lactamase expressed under the control of ldhA promoter fragments of different lengths
3 结论与讨论
本研究从米根霉菌株AS 3.819基因组中克隆了4 种基因ldhA、pdcA、amyA和pgk1的启动子片段,将其重组入构建的启动子探针载体pUKMR中,并以大肠杆菌为宿主,筛选、检测并比较启动子的启动活性。通过比较结果认为,在非底物诱导情况下,Ppgk1和PldhA片段均可以作为强启动子应用于构建米根霉真菌表达载体。而在有合适碳源底物诱导情况下PpdcA和PamyA拥有更高的启动活性,也可作为诱导型强启动子得到应用。ldhA基因的启动子启动活性随着启动子片段长度的增加,启动活性有一定的提高,而在长度为500 bp以上时,其启动活性不再增加。
对于工业生产菌株而言,了解其相关的分子生物学信息对于菌种改造、发酵性能优化、提高其工业应用有着十分重要的意义。通过基因遗传转化技术为基础的代谢工程改造方法是常用的手段,而遗传改造所使用的基因表达载体的构建,除了要选择合适的筛选标记外,适用于宿主菌的高效稳定或者能够进行人为调控的基因启动子也是不可或缺的。在真菌当中,糖化酶基因(amy)和磷酸甘油酸酯激酶基因(pgk)的启动子作为真菌基因的强启动子得到研究与应用[16,24-26],而且由于两种基因的表达类型的不同(amy为诱导型表达,pgk为组成型表达),可以分别在不同的培养条件下达到调节基因表达强度的目的。而丙酮酸脱羧酶基因(pdc)在包括米曲霉在内的丝状真菌中是表达强度最高的基因之一[27],因此其启动子也可作为强启动子用于外源基因在米根霉内的强表达。pdcA、amyA和pgk1这几种基因的启动子片段也分别代表着几种不同类型的启动子,在发酵过程当中,对于不同培养条件其启动基因表达也会做出不同的响应。在米根霉表达载体构建中应用这几种不同类型的启动子米根霉构建不同类型的表达载体,对应着不同的发酵底物,如发酵玉米淀粉(主要碳源底物为淀粉),或者秸秆水解糖(主要含葡萄糖、木糖、阿拉伯糖等单糖)及葡萄糖,能够使得表达载体中的目的基因或者筛选标记基因的表达得到一定的调控,特异性地增强目的基因的表达或者在普通发酵条件下针对性地“关闭”筛选标记基因的表达(降低筛选标记基因的持续表达对宿主米根霉转化子造成的代谢负担)。而本研究中所涉及到的乳酸脱氢酶基因(ldh)启动子目前未有报道对其进行过研究。米根霉是一种重要的有机酸生产菌株,米根霉AS 3.819转化底物碳源为L(+)-乳酸的转化率高,其丙酮酸代谢节点的代谢流量进入乳酸代谢支路的关键酶是乳酸脱氢酶。因此本研究对ldhA基因启动子的启动活性进行了研究,对其不同启动子片段长度的启动能力做了比较,为选择适当的ldhA启动子片段长度提供了参考。另一方面,在本研究中,ldhA启动子活性受碳源底物的影响并不明显,可能说明ldhA基因表达为组成型而非诱导型,但也有可能是因为ldhA基因的表达强度受其他培养底物或者相关代谢途径中的代谢中间物而非碳源底物的影响。其启动子启动基因表达确切的类型仍需进一步研究。
应用启动子探针载体对拥有启动功能的DNA片段进行捕获分离是一种常用的启动子研究手段。目前包括米根霉在内的部分丝状真菌还未建立十分高效稳定且适用范围较广的遗传转化体系,米根霉的主要遗传转化方法也是基于营养缺陷型筛选标记,应用于野生及工业应用的米根霉菌株尚且存在困难[13,28],在这些丝状真菌中进行传统的启动子探针质粒的转化往往得不到理想的效果,给启动子研究带来不便。目前已有研究将丝状真菌的启动子在细菌中启动目的标记基因的表达用以研究其启动表达性能[20,29],且仍有部分启动子保留其启动基因表达的类型,说明这些丝状真菌的基因启动子即使在某些其他的宿主中仍然能有着其基本的启动功能。基于此考虑本研究所构建的启动子探针载体将米根霉的启动子用于在大肠杆菌中启动β-内酰胺酶基因(bla)的表达,并检测了启动子的启动活性。通过这种方法的建立,为米根霉或其他丝状真菌的启动子片段快速简便的捕获分离方法提供参考与借鉴。当然由于启动子在不同的宿主当中的作用应当要考虑宿主的基因表达特性以及目标基因的特异性,筛选所得到的启动子的在米根霉或者丝状真菌中启动基因表达的可靠性以及具体的效果仍需要进一步的验证。在本研究的基础上,将进一步建立并发展米根霉遗传转化方法,并且依靠这种遗传转化方法更深入地分析各种启动子片段在米根霉中的基因启动特性,为米根霉遗传改造工作提供更多理论依据。
参考文献:
[1]BAI Dongmei, LI Shizhong, LEWIS L Z, et al. Enhanced L-(+)-lactic acid production by an adapted strain of Rhizopus oryzae using corncob hydrolysate[J]. Applied Biochemistry and Biotechnology, 2008, 144(1): 79-85.
[2]姜绍通, 郑志 朱羽, 等. 无载体固定化米根霉重复间歇发酵生产L-乳酸[J]. 生物工程学报, 2008, 24(10): 1729-1733.
[3]YAMANE T, TANAKA R. Highly accumulative production of L(+)-lactate from glucose by crystallization fermentation with immobilized Rhizopus oryzae[J]. Journal of Bioscience and Bioengineering, 2013, 115(1): 90-95.
[4]周倩, 赵龙, 蒋雪薇, 等. 一株产生L-乳酸的米根霉[J]. 菌物学报, 2012, 31(6): 956-962.
[5]SAITO K, HASA Y, ABE H. Production of lactic acid from xylose and wheat straw by Rhizopus oryzae[J]. Journal of Bioscience and Bioengineering, 2012, 114(2): 166-169.
[6]杨英歌, 郑之明. 离子注入选育高产L-乳酸的米根霉突变株及其发酵特性研究[J]. 生物技术通报, 2012(7): 152-157.
[7]ZHANG Baohua, YANG Shangtian. Metabolic engineering of Rhizopus oryzae: effects of overexpressing fumR gene on cell growth and fumaric acid biosynthesis from glucose[J]. Process Biochemistry, 2012, 47(12): 2159-2165.
[8]YU Shouzhi, HUANG Di, WEN Jianping, et al. Metabolic profiling of a Rhizopus oryzae fumaric acid production mutant generated by femtosecond laser irradiation[J]. Bioresource Technology, 2012, 114: 610-615.
[9]杨晓娜, 赵昶灵, 李云, 等. 启动子序列克隆和功能分析方法的研究进展[J]. 云南农业大学学报, 2010, 25(2): 283-290.
[10]李珊珊, 迟彦, 李凌飞, 等. 启动子克隆方法研究进展[J]. 中国生物工程杂志, 2005, 25(7): 9-16.
[11]林涛, 黄建忠. 丝状真菌启动子研究进展[J]. 安徽农业科学, 2013, 41(7): 2862-2863; 2865.
[12]徐友强, 马翠卿, 陶飞, 等. 细菌启动子识别及应用研究进展[J]. 生物工程学报, 2010, 26(10): 1393-1403.
[13]MERTENS J A, SKORY C D, IBRAHIM A S. Plasmids for expression of heterologous proteins in Rhizopus oryzae[J]. Archives of Microbiology, 2006, 186: 41-50.
[14]SCHILDE C, WOSTEMEYER J, BURMESTER A. Green fluorescent protein as a reporter for gene expression in the mucoralean fungus Absidia glauca[J]. Archives of Microbiology, 2001, 175: 1-7.
[15]YANAI K, HORIUCHI H, TAKAGI M, et al. Transformation of Rhizopus niveus using bacterial blasticidin S resistance gene as a dominant selectable marker[J]. Current Genetics, 1991, 19: 221-226.
[16]TAKAYA N, YANAI K, HORIUCHI H, et al. Analysis of the 3-phosphoglycerate kinase 2 promoter in Rhizopus niveus[J]. Gene, 1995, 152: 121-125.
[17]李维, 张正义. 以潮霉素B磷酸转移酶基因为遗传标记的启动子探针型载体的构建[J]. 四川大学学报, 1999, 36(4): 736-740.
[18]李维, 张正义. 黄孢原毛平革菌基因启动子的分离与鉴定[J]. 生物工程学报, 2000, 16(5): 599-602.
[19]魏云林, 林连兵, 季秀玲, 等. 低温菌启动子探针质粒的构建[J]. 生物工程学报, 2007, 23(3): 530-534.
[20]李昆, 李松, 牛丹丹, 等. 黑曲霉糖化酶基因启动子功能鉴定[J]. 微生物学杂志, 2008, 28(6): 5-9.
[21]SAMBROOK J, FRITSCH E F, MANIATIS T. Molecular cloning: a laboratory manual[M]. 3rd ed. New York: Cold Spring Harbor Laboratory Press, 2001: 463-470.
[22]张鑫潇. β-内酰胺酶快速检测试剂盒的研究[D]. 郑州: 河南工业大学, 2011.
[23]BRADFORD M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of proteindye binding[J]. Analytical Biochemistry, 1976, 72: 248-254.
[24]HISADA H, SANO M, ISHIDA H, et al. Identification of regulatory elements in the glucoamylase-encoding gene (glaB) promoter from Aspergillus oryzae[J]. Applied Microbiology and Biotechnology, 2013, 97(11): 4951-4956.
[25]HOUGHTON-LARSEN J, PEDERSEN P A. Functional expression of rat adenosine A1 receptor in the dimorphic zygomycete Mucor circinelloides[J]. Applied Microbiology and Biotechnology, 2003, 63: 64-67.
[26]阳辛凤, 郭安平, 孔华, 等. 工业酿酒酵母PGK启动子的克隆与功能分析[J]. 中国农学通报, 2012, 28(6): 178-182.
[27]MAEDA H, SANO M, MARUYAMA Y, et al. Transcriptional analysis of genes for energy catabolism and hydrolytic enzymes in the filamentous fungus Aspergillus oryzae using cDNA microarrays and expressed sequence tags[J]. Applied Microbiology and Biotechnology, 2004, 65(1): 74-83.
[28]SKORY C D. Homologous recombination and double-strand break repair in the transformation of Rhizopus oryzae[J]. Molecular Genetics and Genomics, 2002, 268: 397-406.
[29]汪天虹, 汪浩, 王一彬. 丝状真菌斜卧青霉JUA-10具有启动子功能的DNA片段的克隆与表达[J]. 山东轻工业学院学报: 自然科学版, 2002, 16(1): 61-65.
Cloning and Functional Characterization of Gene Promoters from Rhizopus oryzae AS 3.819
ZHANG Min, JIANG Shaotong*, ZHENG Zhi, PAN Lijun, LI Xingjiang, LUO Shuizhong, WU Xuefeng
(Key Laboratory for Agricultural Products Processing of Anhui Province, School of Biotechnology and Food Engineering, Hefei University of Technology, Hefei 230009, China)
Abstract:In this paper, promoter fragments of four genes, lactate dehydrogenase A (ldhA), pyruvate decarboxylase A (pdcA), glucoamylase A (amyA) and phosphoglycerate kinase 1 (pgk1), from genome DNA of Rhizopus oryzae strain AS 3.819, were amplified and screened in Escherichia coli strain JM109 by using promoter probe vector pUKMR containing β-lactamase gene (bla) as its reporter gene. The results suggested that the four promoter fragments all possessed the ability to drive the expression of the β-lactamase gene. The promoters of ldhA and pgk1 genes possessed high promoting strength even though no inducing substrate was present, while the strength of pdcA and amyA promoters could be enhanced when the suitable carbon source was adopted. The strength of ldhA promoter was enhanced as the fragment length increased until it reached 500 bp. This research provides a quick and easy method to isolate and detect gene promoters form Rhizopus oryzae.
Key words:Rhizopus oryzae; Escherichia coli; promoter probe vector; β-lactamase; lactate dehydrogenase
doi:10.7506/spkx1002-6630-201507014
中图分类号:Q789
文献标志码:A
文章编号:1002-6630(2015)07-0071-08
*通信作者:姜绍通(1954—),男,教授,本科,研究方向为食品发酵工程、大宗农产品资源生物转化加工和油脂加工工程。E-mail:zming19861028@hotmail.com
作者简介:张旻(1986—),男,博士研究生,研究方向为微生物发酵与生物技术。E-mail:zming19861028@126.com
基金项目:国家自然科学基金面上项目(31071636;31171741;31470002);国家自然科学基金青年科学基金项目(31101352)
收稿日期:2014-11-08
猜你喜欢
今日畜牧兽医(2022年10期)2022-12-23
中国现代医药杂志(2020年10期)2020-12-14
医学信息(2017年1期)2017-02-28
分析化学(2016年7期)2016-12-08
国外医药(抗生素分册)(2016年3期)2016-07-12
天津农业科学(2016年4期)2016-04-20
江苏农业科学(2015年5期)2015-10-20
江苏农业科学(2015年8期)2015-09-10
现代检验医学杂志(2015年2期)2015-02-06
现代检验医学杂志(2015年6期)2015-02-06
