
Au-Si6-Au纳米结点电子输运行为的理论计算
2015-03-22 00:53程晓洪张淑华柳福提
华中师范大学学报(自然科学版) 2015年6期
程晓洪, 张淑华, 柳福提,3*
(1.宜宾学院 物理与电子工程学院, 四川 宜宾 644007; 2.宜宾学院 实验与教学资源管理中心, 四川 宜宾 644007;3.宜宾学院 计算物理四川省高校重点实验室, 四川 宜宾 644007)
Au-Si6-Au纳米结点电子输运行为的理论计算
程晓洪1, 张淑华2, 柳福提1,3*
(1.宜宾学院 物理与电子工程学院, 四川 宜宾 644007; 2.宜宾学院 实验与教学资源管理中心, 四川 宜宾 644007;3.宜宾学院 计算物理四川省高校重点实验室, 四川 宜宾 644007)
运用密度泛函理论结合非平衡格林函数的方法,对Si6原子链与两半无限Au(100)-3×3电极耦合构成纳米结点的电子输运行为进行了理论模拟,对结点在不同距离下的电导、结合能进行了计算,结果得到当两电极距离为2.219 nm时,结点结合能较大,结构比较稳定,此时Si-Si平均键长为0.213 nm,Si-Au键长为0.228 nm.对于稳定结构结点,平衡电导为1.093 G0,电子主要通过Si原子的px与py态电子形成的π键进行传输;在-1.2~1.2 V的电压范围内,Si原子链导体具有比较稳定的电导,表现出类似金属的导电特性,其I-V曲线近似为直线关系.
纳米结点; 硅原子链; 电子输运; 第一性原理
连接分子器件的导体必须不断微型化,其极限模型单原子链导体的电子输运特性成为当前大家关注的热点问题.由于其尺寸的限制,它具有许多不同于其块体材料的重要性质.电子传输以弹道输运为主[1],对其导电特性的研究可以进一步扩展人们对介观系统电子输运的理解[2].许多学者从理论上对Ag、Al、Au、Cu、Ir、Na、Pt等金属原子链的电子输运特性进行了深入的研究[3-7],发现电导量子化现象,电导随原子数目的增加发生周期振荡变化的现象.还有研究表明半导体或绝缘体的单原子链的导电性可能比金属原子链还好,如直线C原子链的电导大于Au原子链[8].因此非金属元素及其化合物材料如Ge、Sn、S、AlP、GaAs、CdS、ZnSe、SiC等原子链的电导也得到了极大的关注[9-12].在实验上,人们可以运用力学可控劈裂结(MCBJ)等方法构造各种纳米结点,利用扫描隧穿显微镜(STM)对其电导进行测量.目前,基于密度泛函理论(DFT)结合非平衡格林函数(NEGF)方法是公认为比较精确的处理介观体系电子输运重要方法.鉴于Si低维材料在纳米器件中的重要应用前景,许多研究者对Si团簇[13-14]、纳米线[15-17]、纳米管[18-19]的电子输运性质进行了广泛的研究,为Si纳米线在纳米器件中设计与应用提供了重要参考.为了模拟STM测量纳米级导体电导的实际情形,更好地理解量子传输机理,本文以Si6原子链与Au电极相连构成Au—Si6—Au三明治结构纳米结点,采用DFT结合NEGF的方法对其电子输运行为进行第一性原理模拟计算.
1 计算模型与方法
由于原子链与电极相连构成一个两极模型,其结构包括中央散射区及两半无限电极,其中左、右两半无限电极为理想晶体结构,而中央散射区是由原子链及若干电极层组成.为了尽可能地模拟扫描隧道显微镜工作的实际情况,减少结点处耦合形貌对原子链电子传输的影响,在Si6原子链与Au电极的耦合处增加了5个Au原子金字塔形的耦合结构,让原子链两端Si原子在Au电极的顶位上相互作用,让Si6原子链、左7层电极、右6层电极Au原子相互作用一起构成中央散射区.研究发现在小电压下,有限截面Au电极与周期三维体系电极特别是(100)晶体方向给出的输运特性几乎完全一致[20],所以本文中的电极都选择Au(100)有限截面,同时为了考虑镜像效应,在垂直电子输运方向(z轴)取3×3的超晶胞,计算模型如图1所示.采用DFT[21]+NEGF[22]的方法,以SIESTA程序[23]作为DFT计算平台,获得单粒子Kohn-Sham哈密顿量,运用SMEAGOL[24]程序对连接在两个半无限长Au(100)-3×3电极之间Si原子链的电子输运行为进行完全自洽的计算.

图1 硅原子链纳米结点的结构Fig.1 The structure of the silicon chain nanoscale junctions
在非平衡格林函数方法中,可以根据非周期性开放系统的哈密顿量求解计算模型体系的推迟格林函数GR:

(1)

当施加一定的电压后,通过结点的电流可以通过Landauer-Buttiker[25]公式
(2)
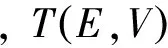
(3)
求出,式(3)中的
有了透射率按(4)式求出体系的平衡电导
(4)
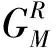
(5)
由密度矩阵可以进一步算出非平衡态下的电子密度,
(6)

在计算中,交换关联能选取Perdew和Zunger[26]所提出的参数化泛函形式进行局域密度(LDA)近似,Au的价电子组态为5d106s1,Si的价电子组态为3s23p2.电极Au原子的价电子用单函数数值基组(SZ)进行展开,而Si原子的价电子用双函数数值基组(DZ),内层芯电子全部采用Troullier-Martins标准模守恒赝势[27]来有效代替库仑势.截断能的大小选取为200Ry,收敛标准为10-4eV.在垂直于电子输运方向的二维布里渊区内分为4个不可约点,布里渊区K点取样为2×2×100.在电荷密度积分计算中,在复平面上沿着半圆选择50个积分点进行计算,而虚轴选择20个能量积分点、费米分布函数选择20个积分点来进行.
2 计算结果与讨论
2.1 结点电导、结合能随距离的变化关系
当结点在某一距离(用dz表示,如图1所示)时,我们先固定两电极的原子,让两金字塔底间的所有原子发生弛豫,进行几何结构的优化,找到能量最低的结构,然后再计算平衡电导与结点的结合能.为了考察结点电导随两电极之间距离变化的关系,我们不断增加两极之间的距离dz,分别计算不同dz下的平衡电导,得到的结果如图2所示.在曲线中电导用方框表示,其值对应左侧纵坐标轴.当dz=2.179 nm时,结点平衡电导为1.308G0(G0=2e2/h,为量子化电导单位,其中e为电子电荷量,h为普朗克常量);随着两极距离的增大,电导有逐渐减小的变化趋势,直至当dz=2.299 nm时,此时电导减小为0.852G0;当dz=2.379 nm时,电导增大到1.181G0,在此距离下,Si链中间两个原子之间的距离变大,即将发生断裂,原子之间的作用情况与之前不同,通过分析透射谱发现此结构结点更有利于电子传输,所以电导反而还略有变大的趋势.但此后随着距离继续增大时,电导快速减小,如当dz=2.419 nm时,电导骤然下降到0.098G0,此时意味着结点内原子耦合很弱,原子链可能已经断裂.从计算结果容易看出,结点平衡电导G随两极距离dz的变化非常明显,在dz变化不到0.3 nm的范围内,电导G却有一个数量级的变化,这充分说明两电极之间的距离对结点的电导有决定性的影响作用,而这也恰好就是STM灵敏度很高的物理机制.在不同的距离下,由于结点处原子间的相互作用,经过充分弛豫后的几何结构不同,电子云空间分布情况发生变化,从而电子通过结点传输的行为也就不完全一样.如当dz=2.219 nm时,Si原子链中的键长依次为0.212、0.215、0.210、0.215、0.212 nm,成对称分布,平均Si-Si键长为0.213 nm,比自由链中的原子键长要小,相互作用较强,使得能级发生移动与展宽.原子链中的成键情况可用Si≡Si-Si≡Si-Si≡Si来简单的表示,正是由于存在Si≡Si三键(共用三个电子对)、Si-Si单键(共用一个电子对)的交替变化,因而出现键长的交替变化.两端Si原子存在未满的悬挂键,因而可与两电极的Au原子的电子相互作用,耦合处Au与Si原子之间的键长dAu-Si=0.228 nm.而随着结点的拉伸,原子链中的键长发生变化,金字塔顶的Au原子与Si原子之间的距离也随之发生改变.当dz=2.419 nm时,dAu-Si=0.229 nm,Si链的平均键长为dSi-Si=0.245 nm,而中央两个Si原子之间的距离长达0.337 nm,相互作用很弱,化学键可能已经断裂,结构如图2中的插图所示,此时结点电导很小.

图2 结点电导、结合能随两极距离的变化关系Fig.2 Conductance and the cohesion energy of junctions as a function of distance
为找到结点的稳定结构,计算了不同dz下结点中央散射区的结合能,ΔE=E(Au-Si6-Au)-E(Si6原子链)-E(Au电极),其中E(Au-Si6-Au)是中央散射区Si6原子链与Au原子的总能量,E(Si6原子链)是孤立Si6原子链的能量,E(Au电极)为孤立Au电极的能量.结果也如图2所示,在曲线中结合能用圆圈表示,其值对应右侧纵坐标轴.从图2中很容易看出,两电极距离dz从2.179 nm逐渐增加到2.459 nm的过程中,当dz=2.219 nm时,结合能比较大,表示此时结点结构比较稳定,我们称这个位置时的结构为稳定结构,此时两侧对顶位的Si-Au键长都为0.228 nm,原子链中Si-Si平均键长为0.213 nm,结点的平衡电导为1.093G0,比其他文献计算得到相同数目原子的Al[2]、Ag[6]等金属原子链的平衡电导值大.
2.2 透射谱与投影态密度
对于稳定结构时的结点,当电压为零时,它的透射率随入射电子能量的变化关系(透射谱)如图3中下半部分所示,从图3中容易看出,在费米面EF(本文中已设置为零)附近有4个明显的透射峰,对应于原子链的分子轨道.最高占居轨道(HOMO)隧穿共振峰位于E-EF=-0.743 eV处,而最低未占据轨道(LUMO)隧穿共振峰位于E-EF=0.109 eV处,它比HOMO峰更靠近费米能级,说明低能量电子主要是通过原子链的LUMO进行传输的.在费米能级处的透射率为1.093,因此平衡电导G=1.093G0.为了进一步分析电子隧穿通道的主要构成,我们计算了Si原子链的投影态密度(PDOS),结果如图3中上半部分所示.由于计算得到投影到Si原子链的px、py的态密度基本上相等,图3中把两者合在一起用px+py来表示.从图3中很容易发现,px+py曲线形状与透射谱基本一致,这说明电子通过结点时,对传输起主要作用的是Si原子的px、py轨道相互作用形成的π键,形成了透射共振的LUMO峰,而s、pz轨道电子局域性较强,对电子传输的贡献较小.
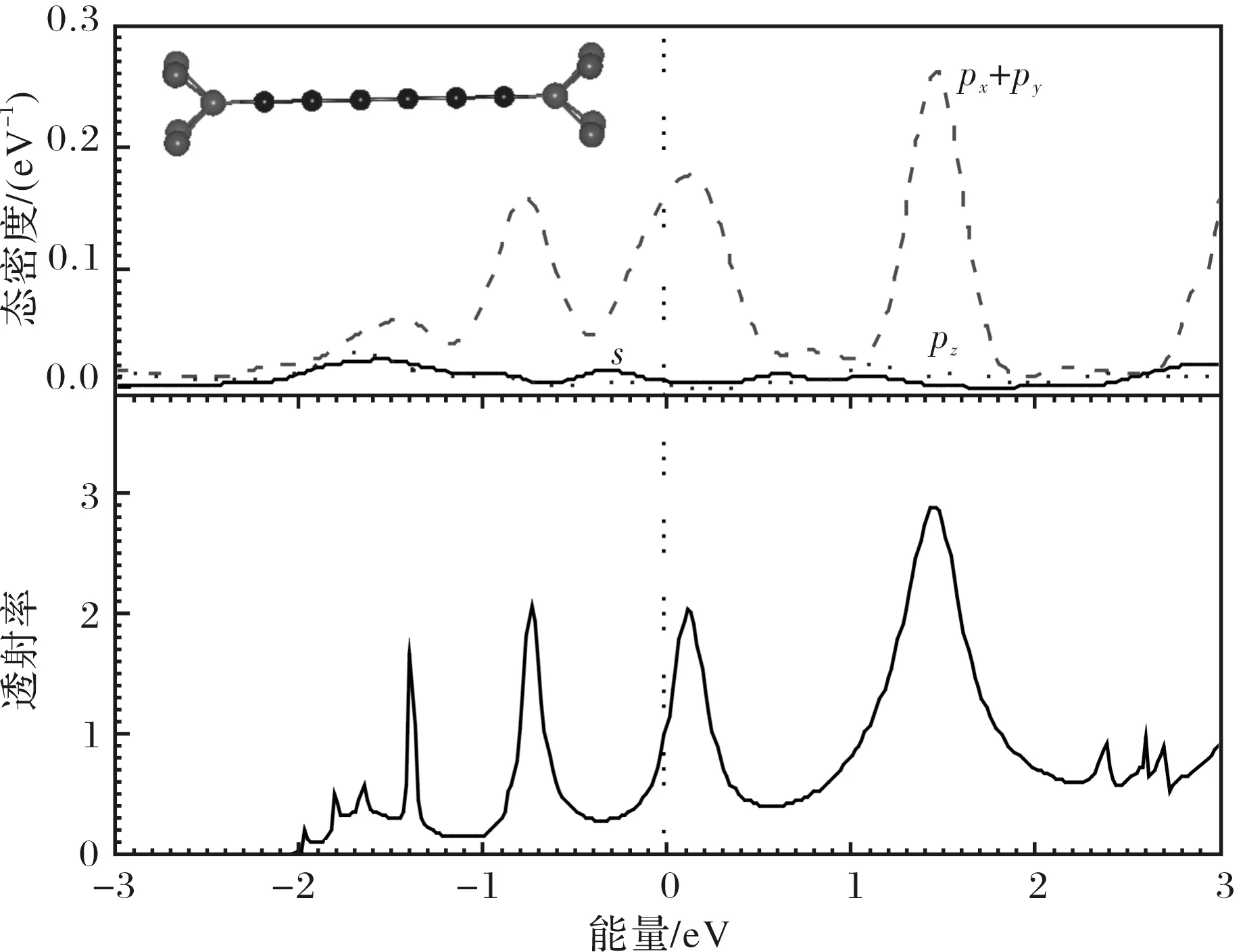
图3 稳定结构结点的态密度与透射谱Fig.3 Density of states and transmission coefficient of junctions as a function of energy in equilibrium position
2.3 电流—电压关系


图4 稳定结构结点的电流随外电压的变化关系Fig.4 The current as a function of bias for junctions in equilibrium position

图5 稳定结构结点不同电压下的透射谱Fig.5 Transmission coefficient of junctions as a function of energy under different bias for junctions in equilibrium position
3 结论
运用密度泛函理论结合非平衡格林函数的方法,对Si6有限长原子链耦合在Au(100)-3×3两电极之间形成纳米结点的电子输运行为进行了第一性原理计算.考察了结点在不同距离下的平衡电导、结合能,结果发现当两极距离dz=2.219 nm时,结点结合能取得极大值,结构最稳定,此时原子链中Si-Si的平均键长为0.213 nm,原子链两端与电极耦合的Si-Au键长为0.228 nm.对于稳定结构的平衡电导为1.093 G0,电子传输主要通过Si原子的px与py轨道电子形成的π键进行的;在-1.2~1.2 V的电压范围内,透射谱形状基本不变,透射共振峰的位置向低能量方向移动,其I-V曲线近似为直线,表现出类似金属的导电特性,说明Si原子链在不同电压下具有比较稳定的电子输运行为.希望本文预测出的电子传输特性能引起实验工作者的兴趣,在实验上得到成功测量,为Si原子链分子器件的设计提供重要参考.
[1] EGAMI Y, AIBA S, HIROSE K, et al. Relationship between the geometric structure and conductance oscillation in nanowires[J]. Journal of Physics: Condensed Matter, 2007, 19(36): 365201(1-10).
[2] THYGESEN K S, JACOBSEN K W. Four-atom period in the conductance of monatomic Al wires [J]. Phys Rev Lett, 2003, 91: 146801(1-4).
[3] SMIT R H M, UNTIEDT C, YANSON A I, et al. Common origin for surface reconstruction and the formation of chains of metal atoms[J]. Phys Rev Lett, 2001, 87: 266102(1-4).
[4] SMIT R H M, UNTIEDT C, RUBIO-BOLLINGER G, et al. Observation of a parity oscillation in the conductance of atomic wires [J]. Phys Rev Lett, 2003, 91: 076805(1-4).
[5] BAHN S R, JACOBSEN K W. Chain formation of metal atoms[J]. Phys Rev Lett, 2001, 87: 266101(1-4).
[6] LEE Y J, BRANDBYGE M, PUSKA M J, et al. Electron transport through monovalent atomic wires[J]. Phys Rev B, 2004, 69: 125409(1-5).
[7] WAN C C, MOZOS J L, TARASCHI G, et al. Quantum transport through atomic wires[J]. Phys Rev Lett, 1997, 71: 419-421.
[8] TONGAY S,SENGER R T, DAG S, et al. A bi nitio electron transport calculations of carbon based string structures[J]. Phys Rev Lett, 2004, 93:136404(1-5).
[9] SENGER R T, TONGAY S, DURGUN E, et al. Atomic chains of group-IV elements and III-V and II-VI binary compounds studied by a first-principles pseudopotential method[J]. Phys Rev B, 2005, 72(7): 075419(1-9).
[10] LANDMAN U, BARNETT R N, SCHERBAKOV A G. et al. Metal-semiconductor nanocontacts: silicon nanowires[J]. Phys Rev Lett, 2000, 85: 1958-1961.
[11] 陈小春, 杨 君, 周艳红, 等. 碳硅链和氮铝链的电子输运特性的第一性原理研究[J].物理学报, 2009, 58(5): 3064-3070.
[12] YU J X, CHEN X R, SANVITO S, et al. Quantum transport of Au-S-S-Au nanoscale junctions[J]. Appl Phys Lett, 2012, 100: 013113(1-4).
[13] ROLAND C, MEUNIER V, LARADE B, et al. Charge transport through small silicon cluster[J]. Phys Rev B, 2002, 66(3): 035332(1-7).
[14] DAI Z X, ZHENG X H, SHI X Q, et al. Effects of contact geometry on transport properties of a Si4cluster[J]. Phys Rev B, 2005, 72(20): 205408(1-9).
[15] LIU F T, CHENG Y, YANG F B, et al. Electron transport through a silicon atomic chain[J]. Chin Phys Lett, 2013, 30(6): 067302(1-4).
[16] LIU F T, CHENG Y, YANG F B, et al. Quantum conductance oscillation in linear monatomic silicon chains[J]. Physica E, 2014, 56: 96-101.
[17] MOZOS J L,WAN C C,TARASCHI G, et al. Quantized conductance of Si atomic wires[J]. Phys Rev B, 1997, 56: 4351-4354.
[18] DONG J C, LI H, SUN F W, et al. Theoretical study of the properties of Si nanowire electronic devices [J]. J Phys Chem C, 2011, 115(28): 13901-13906.
[19] SVIZHENKO A, LEU P W, CHO K. Effect of growth orientation and surface roughness on electron transport in silicon nanowires[J]. Phys Rev B, 2007, 75(12): 125417(1-7).
[20] KE S H, BARANGER H U, YANG W. Contact atomic structure and electron transport through molecules[J]. The Journal of Chemical Physics, 2005, 122(7): 074704(1-9).
[21] KOHN W, SHAM L J. Self-consistent equations including exchange and correlation effects[J]. Phys Rev B, 1965, 140:1133-1138.
[22] DATTA S. Electronic Transport in Mesoscopic Systems[M]. Cambridge: Cambridge University Press,1995:19.
[23] SOLER J M,ARTACHO E,GALE J D, et al. The SIESTA method for ab initio order-N materials simulation[J]. Phys Condens Matt, 2002, 14:2745(1-22).
[24] ROCHA A R,GARCIA-SUAREZ V M,BAILEY S, et al. Spin and molecular electronics in atomically generated orbital landscapes[J]. Phys Rev B, 2006, 73:085414(1-22).
[25] BÜTTKIER M, IMRY Y, LANDAUER R, et al. Generalized many-channel conductance formula with application to small rings[J]. Phys Rev B, 1985, 31(10): 6207-6215.
[26] PERDEW J P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas[J]. Phys Rev B, 1986, 33:8822-8824.
[27] TROULLIER N, MARTINS J L. Efficient pseudopotentials for plane-wave calculations[J]. Phys Rev B, 1991, 43:1993-2006.
[28] 柳福提, 程 艳, 羊富彬, 等. Au-Si-Au 结点电子输运性质的第一性原理计算[J].物理学报, 2013, 62(10): 107401(1-6).
Theoretical calculation of electron transport behavior of Au-Si6-Au nanoscale junctions
CHENG Xiaohong1, ZHANG Shuhua2, LIU Futi1,3
(1.School of Physics and Electronic Engineering of Yibin University, Yibin, Sichuan 644007;2.Centre for Experimental and Teaching Resource Management of Yibin University, Yibin, Sichuan 644007;3.Computational Physics Key Laboratory of Sichuan Province of Yibin university, Yibin, Sichuan 644007)
Electron transport behavior of Au-Si6-Au nanoscale junctions, which consists of six silicon atoms sandwiched between two semi-infinite Au(100)-3×3 electrodes, were investigated with combination of density functional theory and the non-equilibrium green’s function method from first principles. We calculated conductance and the binding energy in different distance. The results show that the binding energy of junctions is near to maximum and the structure is stable whendz= 2.219 nm. The corresponding average length of Si-Si and Si-Au bond is 0.213 nm and 0.228 nm, respectively. Besised, the equilibrium conductance is 1.093G0. The electrons transmit mainly through the tunneling channel of bonds formed bypxandpyorbital electrons of Si atoms. In voltage range from -1.2 to 1.2 V, silicon atomic chain conductor has a stable electron transport behavior, and theI-Vcurve of the junctions show approximate linear feature similar to the metal conductor.
nanoscale junctions; silicon atomic chain; electron transport; first principles
2015-05-11.
四川省自然科学基金项目(13ZB0207);宜宾学院科研基金项目(2012S12).
1000-1190(2015)06-0861-06
O469
A
*通讯联系人. E-mail: futiliu@163.com.
猜你喜欢
大学物理(2022年9期)2022-09-28
物理通报(2020年7期)2020-07-01
青岛大学学报(工程技术版)(2019年2期)2019-09-10
电子制作(2018年14期)2018-08-21
电测与仪表(2016年20期)2016-04-11
枣庄学院学报(2015年5期)2016-01-09
西南医科大学学报(2016年4期)2016-01-03
中学化学(2015年8期)2015-12-29
原子与分子物理学报(2015年3期)2015-11-24
中国药业(2014年21期)2014-05-26
