
Theoretical study of BTF/TNA cocrystal:Effects of hydrostatic pressure and temperature
2015-11-01 07:13PengyuanCHENLinZHANGShunguanZHUGuangbinCHENG
Defence Technology 2015年2期
Peng-yuan CHEN,Lin ZHANG*,Shun-guan ZHU,Guang-bin CHENG
School of Chemical Engineering,Nanjing University of Science and Technology,Nanjing 210094,Jiangsu,China
Theoretical study of BTF/TNA cocrystal:Effects of hydrostatic pressure and temperature
Peng-yuan CHEN,Lin ZHANG*,Shun-guan ZHU,Guang-bin CHENG
School of Chemical Engineering,Nanjing University of Science and Technology,Nanjing 210094,Jiangsu,China
Cocrystallization is a promising technique for the design and preparation of new explosives,and the stability of cocrystal is highly concerned by the researchers.In order to make a better understanding of the behavior of cocrystal under the extreme conditions,DFT(density functional theory)calculation is performed to investigate the effect of hydrostatic pressure on geometrical and electronic structures of the cocrystal BTF(benzotrifuroxan)/TNA(2,4,6-trinitroaniline).When the hydrostatic pressure is applied,the lattice constants,volume,density and total energy change gradually except at the pressures of 40 GPa and 79-83 GPa.It is noteworthy that new chemical bonds form when the pressure is up to 83 GPa.The band gap of the cocrystal becomes smaller when the pressure is applied,and finally the cocrystal shows a characteristic of metal. The mechanical property of cocrystal is calculated by MD(molecular dynamics)simulation.The results show that the cocrystal has a better ductibility at low temperature,and has the best tenacity at 295 K.
Cocrystal;BTF/TNA;DFT;High pressure;Crystal structure
1.Introduction
Cocrystal is defined as a new crystal with distinct solidstate properties created by combining two or more neutral species in a definite ratio[1,2].Cocrystallization is an effective approach to alter the dissolution rate,thermal stability and bioavailability of pharmaceuticals in pharmaceutical industry[3-5].Its great success in pharmaceutical industry has attracted much attention in other fields,such as optical,semiconducting materials and energetic materials[6-8].Cocrystallization can modify the performance of energetic material at molecular level compared with coating,crystal morphology modification and other traditional methods[9-11].It is feasible to tailor the different molecules to get the definite performance according to some rules with cocrystallizationmethod,which has been verified in pharmaceutical fields. Seventeen cocrystals of TNT(2,4,6-trinitrotoluene)were reported and the safety performance has been greatly enhanced[12].However,the energy is diluted because of the introduction of non-energetic materials.Bolton et al.[13]used the solvent evaporation method to prepare the cocrystal of TNT/ CL-20(2,4,6,8,10,12-hexanitrohexaazaisowurtzitane).The cocrystal has a poorer detonation performance but a reduced sensitivity compared with pure CL-20.The cocrystal of HMX(1,3,5,7-tetranitro-1,3,5,7-tetrazocane)/CL-20 is predicted to be more powerful than HMX[14].Nevertheless,the sensitivity of the cocrystal is similar to that of HMX.Millar et al.[15]changed the physicochemical properties of CL-20 by cocrystallizing CL-20 with four solvents.It is notable that the CL-20 molecule adopts different conformations before and after desolvation,and this may provide a highly-selective means of polymorph screening.
When a detonation takes place,the high velocity shock waves can result in very high temperatures and pressures[16],which may lead to a phase transition of energetic material to form new polymorphs or decomposition reactions[17,18].As a result,the crystal structure would deform.It is well known that the sensitivity and detonation performance of the explosive are intimately bound up with the crystal packing. Therefore it is necessary to investigate the behavior of explosives under the extreme conditions.In recent years,a lot of efforts have been laid both in experiment and theoretical calculation in this respect.Diamond anvil cell,which can generate a pressure of hundreds of GPa,was used to study the changes of materials at high pressure,such as cell parameters,phase transition,and so on[19,20].Compared with the experiment,theoretical calculations show much more details to help us to understand the changing mechanism.Microscopic changes of geometrical structure and electronic distribution can be studied by theoretical calculation.To date,a lot of widely used explosives,such as HMX,CL-20[21],RDX(hexahydro-1,3,5-trinitro-1,3,5-triazine)[22],andsoon[23-27],at different hydrostatic pressures were studied using the computational methods.As for the cocrystal explosives,it is of great importance to consider their stability at high pressure.However,the formation and decomposition mechanism of the cocrystal explosive is not clear,and its behavior under theextremeconditionsisnotmentioned.Furthermore,whether the molecular interactions are destroyed in the process of detonation is concerned by the researchers,especially considering the advantages of cocrystal explosives that have been demonstrated.However,to our knowledge,no work has been reported on the study of the behavior of cocrystal explosive at high pressure.
http://dx.doi.org/10.1016/j.dt.2015.01.003
2214-9147/Copyright©2015,China Ordnance Society.Production and hosting by Elsevier B.V.All rights reserved.
By the previous study,BTF can be cocrystallized with a series of explosives,including TNA,TNT,CL-20,and so on. Generally,the sensitivity of the cocrystal is between those of both BTF and TNA.However,the sensitivity of BTF/TNA[28]is higher than those of BTF and TNA.Hence,BTF/ TNA is more worthy of theoretical investigation.For comparison,a blank test was also performed in the absence of the other partner of the couple.As we all know,the mechanical property is an important indicator of the explosive property,and the shear modulus is related to the sensitivity of explosive.Therefore,the mechanical properties of the cocrystal at different temperatures were calculated by MD simulation.
2.Computational methods
The calculations in this study were performed within the framework of DFT based on CASTEP code[29].The LDA functional proposed by Ceperley and Alder[30]and parameterized by Perdew and Zunder[31],named CA-PZ,was employed.The core electrons in each atomic species were replaced with the norm-conserved pseudopotentials.The primitive cell was relaxed by using the BFGS method.The cutoff energy of plane wave was set to 600 eV.Brillouin zone sampling was performed by using the Monkhost-Pack scheme with a k-point grid of 2×2×1.The values of the kinetic energy cutoff and the k-point grid were determined to ensure the convergence of total energy.In the geometry relaxation,the total energy of the system was less than 2.0×10-5eV,the residual force was less than 0.05 eV/Å,the displacement of atoms was less than 0.002 Å,and the residual bulk stress was less than 0.1 GPa.
All MD simulations were carried out in an isothermalisobaric ensemble(NPT)with the COMPASS[32]force field in Discover code.The unit cell was taken from single X-ray diffraction.A supercell consisting of 3×3×2 unit cells was prepared and put into the periodic simulation cell.Each cell contains 72 BTF molecules,72 TNA molecules and 2376 atoms in total.Temperature was controlled by using the stochastic collision method proposed by Andersen[33].The temperatures were set to 195 K,245 K,295 K,345 K and 395 K.The initial velocities were set using the Maxwell-Boltzmann profiles at given temperatures.The velocity Verlet time integration method was used with a time step of 1 fs.The Verlet leapfrog scheme was used to integrate the equations of motion.The total simulation time was set to 2 ns,the equilibration was achieved in the first 1 ns,and the data was collected in the next 1 ns.
3.Results and discussion
3.1.Crystal structures and properties
LDA and GGA often give contradictory results when the crystal structure is optimized[16].Therefore,in order to benchmark the performances of LDA and GGA,both those functionals were applied to the cocrystal of BTF/TNA as a test.As shown in Table 1.After accomplishing total energy minimization for each unit cell,the optimized lattice parameters and the experimental results shows that the LDA results are in well agreement with the experimental ones compared to the GGA results.As for LDA functional,the relative errors of the lattice constants a,b,c,and β are-3.06%,-0.66%,+1.05%,and+0.07%,respectively.The little difference between the calculated and experimental lattice parameters demonstrates that the method and the parameters used in this calculation are reliable.However,the difference may be due to the intermolecular interactions occurring in the crystal lattice,which are not well described by DFT[34].
The experimental crystallization and structure of BTF/TNA cocrystal are presented in Figs.1 and 2.Each unit cell has four BTF molecules and four TNA molecules.
The relaxed lattice constants of the cocrystal at different hydrostatic pressures are presented in Fig.3.On the whole,thesmaller the compression ratio is.For example,in the region of 0-10 GPa,the compressions of a,b and c are 9.50%,6.30% and 9.03%,respectively,which are much larger than 1.80%,1.03%and 0.92%in the region of 50-60 GPa.In the lowpressure zone,the distance between the molecules is far,so the intermolecular repulsion is not large.Therefore,the crystal is easily compressed compared to chemical bond.On the contrary,in the high-pressure zone,the intermolecular repulsion is much larger,and the crystal is difficult to be compressed.As is evident,the compressibilities along three directions are different,which indicates that the compressibility of the crystal is anisotropic.As the pressure goes up,the compressibility in each direction converges.Additionally,the crystal symmetry maintains the same monocline space group P21/c under all hydrostatic compressions.

Table 1 The experimental and calculated lattice parameters of BTF/TNA cocrystal.a
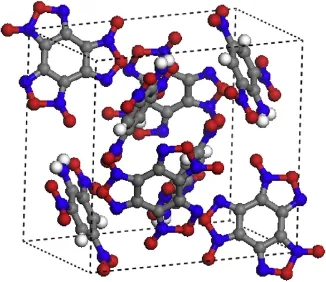
Fig.1.The experimental crystallization of BTF/TNA cocrystal.

Fig.2.The molecular structure and atomic labeling of BTF/TNA cocrystal.

Fig.3.The optimized lattice constants(a,b and c)as a function of pressure.
The volume,density,and total energy of optimized unit cell as a function of pressure are depicted in Fig.4.With the inlattice constants reduce with the increase in pressure.It is worth noting that all the lattice constants increase abnormally at 40 GPa,while in the region of 79-83 GPa,b and c increase again,but a decreases sharply.It can be deduced that the structural transformation may happen in those regions.Obviously,the largest compression of the unit cell takes place at low pressure below 10 GPa.The higher the pressure is,the crease in pressure,the volume of the unit cell decreases constantly.Obviously,like the lattice parameters,the unit cell volume has the largest compression ratio at low pressure.For instance,the volume is reduced by 3.69%in the region of 50-60 GPa,which is much smaller than that(22.86%)in the region of 10-20 GPa.The change in energy is very similar to that in density,and they all increase with the increase in pressure.The energy and density decrease significantly at the pressure of 40 GPa.As shown in Fig.5,the distance of N1O1···H7A decreases gradually with the increase in pressure.However,at 40 GPa,the length of hydrogen bond is 1.668 Å,which is smaller than 1.790 Å at 30 GPa or 1.764 Å at 50 GPa.Hence,the strength of hydrogen bond increases greatly due to the decrease in bond lengths.The crystal becomes more stable and the total energy decreases.The density and total energy raise linearly with the increase in pressure below 40 GPa.The higher total energy means that the more the explosive releases blasting energy during the explosion,the more powerful the explosive is[16].Meanwhile,the pressure-induced density enlargement would enhance the detonation performance.As a result,they show the same changing tendency with the increase in pressure.For pure BTF crystal,the lattice constants,bond lengths and some otherparameters of BTF change gradually with the increase in pressure.No isomerization or other particular changes were observed even at the applied pressure of 100 GPa.

Fig.4.The volume,density,and total energy of optimized unit cell as a function of pressure.
3.2.Molecule structure
Both the unit cell and the molecular geometry are affected by the external pressure.Some main geometrical parameters, including bond length and torsion angle,are presented to illuminate the effect of hydrostatic pressure.

Fig.5.Variation of the hydrogen bond length at different pressures.
As shown in Fig.6,the imposed compression mostly squeezes out the intermolecular space and causes only a little change in intramolecular geometry in the low-pressure zone. Some bonds,such as N1-O2,C1-C2,C7-C8,C8-N8,C8-C9 and C7-N7,shorten gradually and obviously,while some other bonds,such as N2-O2 and N1-O1,change a little bit.This indicates that an endocyclic ring is more likely to be compressed.N1-O2,N9-O9,N1-O2 and N9-O9 shorten abruptly at 40 GPa.Conversely,C1-C2 and N2-O2 lengthen all of a sudden.At 79 GPa,N1-O2 shorten while N2-O2 lengthen.This indicates that N2-O2 may be broken when the explosive decomposes.Simultaneously,in the TNA molecule,the C-NO2bonds adjacent to the amino lengthen suddenly,indicating that the C-NO2bond is the trigger bond of TNA,which is consistent with the previous study[35].
ThetorsionanglesofC1-C6-C5-N5and C9-C8-C7-N7 are 179.208°and 177.444°,respectively,at 0 GPa,which are close to 180°(Fig.7).Therefore,the fivemembered furoxan ring and the six-membered ring in BTF are almost on one plane,amino and benzene rings in TNA are also on the same plan.In the region of 0-30 GPa,the torsion angle of C1-C6-C5-N5 is sharply decreased to 144.528°because of the effect of pressure,that is to say,the fivemembered furoxan ring forms an angle with the benzene ring.On the other hand,the torsion angle of C9-C8-C7-N7 decreases significantly in the region of 0-20 GPa,then it increases at the following 10 GPa.Following this,the torsion angle decreases significantly at 40 GPa,and then the angle maintains around 170°.When the pressure gets higher,the distance between the molecules becomes smaller.So the distance between O1 and H7A decreases from 2.279 Å to 1.790 Å.Consequently,they form a strong hydrogen bonding. With the attraction of O1,H7A moves from the original position.As a result,C7-N7 forms a torsion angle with the benzene ring.After that,the hydrogen bonding becomes stable so that the angle does not show obvious change.

Fig.6.Variation of the bond lengths at different pressures.

Fig.7.Variation of the torsion angles at different pressures.
Fig.8 shows the perspective views of one layer of BTF/ TNA cocrystal unit cell at the selected pressures.When the pressure is higher than 82 GPa,something unreasonable changes happen in the geometry.At 83 GPa,O in the nitro group of TNA forms a chemical bond with the C in the benzene ring of the adjacent TNA molecule.More chemical bonds can be found in the unit cell among TNA molecules at 100 GPa.The same situation was also found in TNA crystal. When the pressure reaches to 64 GPa,O in the nitro group forms a chemical bond with C in the benzene ring.
3.3.Electronic structure
Density of state(DOS)analysis is useful to understand better the change of electronic structure caused by external pressure[36].The calculated total density of states(TDOS)of BTF/TNA cocrystal at different pressures is shown in Fig.9. Fermi level is plotted by the dash line.The main characteristics of DOS are summarized as follows:(1)The top of valence band and the bottom of conduction band are mainly due to p orbit,indicating that p orbit plays a key role in chemical reaction;(2)When the pressure is low,the curves of DOS are characterized by obvious peaks,but the peaks widen gradually with the increase in pressure,which means that the band splitting and band dispersion are accompanied by a broadening of DOS;and(3)Moreover,the conduction bands have a tendency of shifting to the lower energy.This leads to the band gap be smaller,and the crystal shows the characteristic of metal[37].
The energy gap between the highest occupied crystal orbit(HOCO)and lowest unoccupied crystal orbit(LUCO)is a main parameter to characterize the electronic structure of a solid[17].It can be seen from Fig.10 that the band gap decreases from 2.019 eV to 1.134 eV in the region of 0-30 GPa. This is because of the decrease in intermolecular space under compression which leads to an increase in the overlap of different groups of bands and hence increases the charge overlap and delocalization in the system.The tendency of band gap is similar to that of volume of the bulk.The band gap shows no obvious changes in the region of 30-39 GPa. Because of the structural transformation,the band gap is increased to 1.432 eV at 40 GPa.The band gap decreases gradually in the region of 41-78 GPa.However,the reduction rate is much smaller than that of it in the region of 0-30 GPa. At 79 GPa,the band gap decreases suddenly with the change of bond length.In the region of 80-82 GPa,the band gap rises perpendicularly.Then the band gap decreases again at 83 GPa,which is a turning point to form the unexpected chemical bonds.The formation of the new chemical bonds results in the enhancement of the delocalization of the crystal.In another word,the electron is more likely to transfer and the band gap reduces.Compared with the lattice constants and geometrical parameters,the band gap is more sensitive to pressure.According to the principle of easiest transition(PET)[33],the smaller the band gap is,the more easily the electron transits from the occupied valence band to the empty conduction band. That means the explosive is more likely to detonate.Therefore,the BTF/TNA cocrystal has a higher sensitivity when the external pressure is applied.
3.4.MD simulation results

Fig.8.Perspective views of one layer of BTF/TNA cocrystal unit cell and BTF molecules are omitted for brevity.

Fig.9.Calculated total DOS of BTF/TNA cocrystal at different pressures.
BTF/TNA cocrystal crystallizes in a monoclinic P21/c space group and contains 8 molecules per unit cell.Since the data of single X-ray diffraction were collected at 135 K,we carried out the calculation at 135 K to confirm the applicability and accuracy of the method and parameters applied in the latter calculations.It can be known from Table 2 that The errors among the optimized lattice constants a,b,c and the experimentalvaluesare+0.61%, +0.60, +0.60%, respectively,after the system reached to the equilibrium state. The result shows that the method and the parameters are reliable.
The lattice parameters at different temperatures were calculated and presented in Table 2.It is obvious that the lattice parameters rise with the increase in temperature.

Fig.10.Band gaps of BTF/TNA cocrystal at different pressures.

Table 2 The experimental and calculated lattice parameters at different temperatures.a
The ratio of stress to strain,which calls elastic modulus,is a measure of rigidity.The larger the elastic modulus is,the stronger its rigidity is.Poisson's ratio is connected with the plastic property.Higher value of Poisson's ratio means stronger plastic property.Cauchy pressure(C12-C44)is often used to evaluate the ductibility and brittleness of a material.Usually,the Cauchy pressure of ductile material is positive ductile material while the Cauchy pressure of brittle material is negative.The tenacity of a material can be evaluated by the ratio of bulk modulus to shear modulus(K/G).Usually,the greater the value of K/G is,the better tenacity the material possesses[38,39].
Table 3 lists the isotropic mechanical properties of the BTF/ TNA cocrystal at different temperatures,including tensile modulus,Poisson's ratio,bulk modulus,and so on.Obviously,the values of C11,C22and C33are larger than those of other elastic moduli,indicating that more stress is needed to create the same deformation.However,the other elastic moduli are much smaller,which suggests that the BTF/TNA cocrystal is anisotropic.All the elastic moduli decrease with the increase in temperature.This is consistent with the general law of mechanical properties,that is to say,the higher the temperature is,the weaker rigidity and brittleness the material has.The explosive with a larger shear modulus is considered to be more sensitive than those with a smaller one.However,in this study,the shear modulus of the cocrystal at higher temperature is smaller than that of it at low temperature,which is not consistent with the fact that an explosive becomes more sensitive when temperature increases.This may be because of the fact that the factors which can influence the sensitivity and shear modulus are less important in this situation.Poisson'sratio is almost unchanged in all the temperature regions,indicating that the cocrystal has an inherent plasticity which is not easily affected by temperature.Furthermore,Cauchy pressure lowers with the increase in temperature,which infers the cocrystal has a better ductibility at low temperature.K/G reaches to its largest value at 295 K,which illustrates that the BTF/TNA cocrystal has the best tenacity at 295 K.

Table 3 Tensile modulus(E),Poisson's ratio(ν),bulk modulus(K),shear modulus(G),Cauchy pressure(C12-C44)and quotient K/G at different temperatures(K)for BTF/TNA cocrystal
4.Conclusions
In this study,the theoretical simulations were performed to investigate the behaviors of cocrystal under the extreme conditions.The results show that,when the hydrostatic pressure is applied,the lattice constants,volume,density and total energy change gradually except at 40 GPa and 79-83 GPa.A new chemical bond is found when the pressure is up to 83 GPa. Meanwhile,with the pressure augments,the electron transfers and the band gap decreases significantly.That is to say,the sensitivity of cocrystal becomes higher when the hydrostatic pressure rises.The MD simulation suggests that the cocrystal has a better ductibility at low temperature,and has the best tenacity at 295 K.
Acknowledgments
The authors gratefully acknowledge the support of the National Natural Science Foundation of China(Grant No. 61106078).
[1]Guo CY,Zhang HB,Wang XC.Crystal structure and explosive performanceofanewCL-20/caprolactamcocrystal.JMolStruct 2013;1048:267-73.
[2]Landenberger KB,Matzger AJ.Cocrystals of 1,3,5,7-Tetranitro-1,3,5,7-tetrazacyclooctane(HMX).Cryst Growth Des 2012;12(7):3603-9.
[3]Van L,Janan J,Mary KS.High-throughput 96-well solvent mediated sonic blending synthesis and on-plate solid/solution stability characterizationofpharmaceuticalcocrystals.IntJPharm 2013;441(1-2):356-64.
[4]Mohammad AM,Amjad A,Sitaram PV.Hansen solubility parameter as a tool to predict cocrystal formation.Int J Pharm 2011;407(1-2):63-71.
[5]Jonathan WS.The role of co-crystals in pharmaceutical design.Trends Pharmacol Sci 2013;34(3):185-93.
[6]Landenberger KB,Bolton O,Matzger AJ.Two isostructural explosive cocrystals with significantly different thermodynamic stabilities.Angew Chem Int Ed Engl 2013;52(25):6468-71.
[7]Choi EY,Jazbinsek M,Lee SH.Co-crystal structure selection of nonlinear optical analogue polyenes.CrystEngComm 2012;14(13):4306.
[8]Yang ZW,Li HZ,Zhou XQ,Zhang CY.Characterization and properties of a Novel energetic-energetic cocrystal explosive composed of HNIW and BTF.Cryst Growth Des 2012;12(11):5155-8.
[9]Jung JW,Kim KJ.Effect of supersaturation on the morphology of coated surface in coating by solution crystallization.Ind Eng Chem Res 2011;50(6):3475-82.
[10]Tang YX,Yang HW,Cheng GB.Synthesis and characterization of a stable,catenated N11 energetic salt.Angew Chem Int Ed Engl 2013;52(18):4875-7.
[11]Zhang YQ,Guo Y,Joo YH.3,4,5-Trinitropyrazole-based energetic salts. Chemistry 2010;16(35):10778-84.
[12]Landenberger KB,Matzger AJ.Cocrystal engineering of a prototype energetic material:supramolecular chemistry of 2,4,6-Trinitrotoluene. Cryst Growth Des 2012;10(12):5341-7.
[13]Bolton O,Matzger AJ.Improved stability and smart-material functionality realized in an energetic cocrystal.Angew Chem Int Ed Engl 2011;50(38):8960-3.
[14]Bolton O,Simke LR,Matzger AJ.High power explosive with good sensitivity:a 2:1 cocrystal of CL-20:HMX.Cryst Growth&Des 2012;12(9):4311-4.
[15]Millar DIA,Allan DR,Lennie AR.Crystal engineering of energetic materials:co-crystals of CL-20.CrystEngComm 2012;14(10):3742.
[16]Liu Y,Gong XD,Wang LJ,Wang GX.Effect of hydrostatic compression on structure and properties of 2-diazo-4,6-dinitrophenol crystal:density functional theory studies.J Phys Chem C 2011;115(23):11738-48.
[17]Liu Y,Gong XD,Wang LJ,Wang GX.First-principle studies on the pressure-induced structural changes in energetic ionic salt 3-Azido-1,2,4-triazolium nitrate crystal.J Phys Chem C 2012;116(30):16144-53.
[18]Wang F,Du HC,Zhang JY,Gong XD.First-principle study on highpressure behavior of crystalline polyazido-1,3,5-triazine.J Phys Chem C 2012;116:6745-53.
[19]Davidson AJ,Oswald IDH,Francis DJ,Lennie AR.Explosives under pressure-the crystal structure of γ-RDX as determined by high-pressure X-ray and neutron diffraction.CrystEngComm 2008;10(2):162.
[20]Li SR,Li Q,Wang K,Tan X.Pressure-induced phase transition in guanidiniumperchlorate:asupramolecularstructuredirectedby hydrogen bonding and electrostatic interactions.J Phys Chem B 2012;115(41):11816-22.
[21]Byrd EFC,Rice BM.Ab initio study of compressed 1,3,5,7-Tetranitro-1,3,5,7-tetraazacyclooctane(HMX),cyclotrimethylenetrinitramine(RDX),2,4,6,8,10,12-Hexanitrohexaazaisowurzitane(CL-20),2,4,6-trinitro-1,3,5-benzenetriamine(TATB),and pentaerythritol tetranitrate(PETN).J Phys Chem C 2007;111(6):2787-96.
[22]Patterson JE,Dreger ZA,Gupta YM.Shock wave-induced transition in RDX single crystals.J Phys Chem B 2007;111(37):10897-904.
[23]Liu Y,Du HC,Wang GX,Gong XD.Comparative theoretical studies of high pressure effect on polymorph I of 2,2′,4,4′,6,6′-hexanitroazobenzene crystal.Struct Chem 2012;23(5):1631-42.
[24]Liu Y,Wang GX,Gong XD.2,4-Diazido-5-iodo-pyrimidine crystal under high pressure:a comparison of DFT and DFT-D studies.Comput Theor Chem 2012;1000:60-9.
[25]Zerilli FJ,Kuklja MM.First principles calculation of the mechanical compression of two organic molecular crystals.J Phys Chem A 2006;110(15):5173-9.
[26]Ciezak JA,Jenkins TA,Liu ZX,Hemley RJ.High-pressure vibrational spectroscopyofenergeticmaterials:hexahydro-1,3,5-trinitro-1,3,5-triazine.J Phys Chem A 2007;111(1):59-63.
[27]Qiu L,Zhu WH,Xiao JJ,Xiao HM.Theoretical studies of solid bicyclo-HMX:effects of hydrostatic pressure and temperature.J Phys Chem B 2008;112(13):3882-93.
[28]Zhang HB,Guo CY,Wang XC,Xu JJ.Five energetic cocrystals of BTF by intermolecular hydrogen bond and π-stacking interactions.Cryst Growth&Des 2013;13(2):679-87.
[29]Clark SJ,Segall MD,Pickard CJ,Hasnip PJ.First principles methods using CASTEP.Z Fur Kristallogr 2005;220(5-6):567-70.
[30]Ceperley DM,Alder BJ.Ground state of the electron gas by a stochastic method.Phys Rev Lett 1980;45(7):566-9.
[31]Perdew JP,Burke K,Ernzerhof M.Self-interaction correction to densityfunctional approximations for many-electron systems.Phys Rev B 1981;23(10):5048-79.
[32]Sun H.An ab initio force-field optimized for condensed-phase applicationsoverview with details on alkane and benzene compounds.J Phys Chem B 1998;102:7338-64.
[33]Andersen HC.Molecular dynamics simulations at constant pressure and/ or temperature.J Chem Phys 1980;72:2374-83.
[34]Zhu WH,Zhang XW,Wei T,Xiao HM.First-principles study of crystallinemono-amino-2,4,6-trinitrobenzene,1,3-diamino-2,4,6-trinitrobenzene,and 1,3,5-triamino-2,4,6-trinitrobenzene.J Mol Struct Theochem 2009;900:84-9.
[35]Rice BM,Sahu S,Owens FJ.Density functional calculations of bond dissociation energies for NO2scission in some nitroaromatic molecules.J Mol Struct 2002;583:69-72.
[36]Zhu WH,Xiao HM.First-principles study of electronic,absorption,and thermodynamic properties of crystalline styphnic acid and its metal salts. J Phys Chem B 2009;113(30):10315-21.
[37]Zhu WH,Shi CH,Xiao HM.Density functional theory study of highpressure behavior of crystalline hexanitrostilbene.J Mol Struct Theochem 2009;910:48-153.
[38]Ma XF,Zhao F,Ji GF,Zhu WH.Computational study of structure and performance of four constituents HMX-based composite material.J Mol Struct Theochem 2008;851(1-3):22-9.
[39]Xiao JJ,Wang WR,Chen J,Ji GF.Study on structure,sensitivity and mechanical properties of HMX and HMX-based PBXs with molecular dynamics simulation.Comput Theor Chem 2012;999:21-7.
17 November 2014;revised 18 January 2015;accepted 19 January 2015
Available online 17 March 2015
.Tel./fax:+86 2584315856.
E-mail address:zhangl@mail.njust.edu.cn(L.ZHANG).
Peer review under responsibility of China Ordnance Society.
Copyright©2015,China Ordnance Society.Production and hosting by Elsevier B.V.All rights reserved.

- Defence Technology的其它文章
- Inspection of aluminum alloys by a multi-frequency eddy current method
- 2D and 3D milled surface roughness of high volume fraction SiCp/Al composites
- Determination of penetration depth at high velocity impact using finite element method and artificial neural network tools
- Microstructure and pitting corrosion resistance of AA2219 Al-Cu alloy friction stir welds-Effect of tool profile
- Friction stir surfacing of cast A356 aluminium-silicon alloy with boron carbide and molybdenum disulphide powders
- Variation of chemical composition of high strength low alloy steels with different groove sizes in multi-pass conventional and pulsed current gas metal arc weld depositions