
氧化还原引发制备乙氧酰胺苯甲酯分子印迹微球及其表征
2015-11-29 01:30李兆周李智丽陈秀金李道敏高红丽侯玉泽李松彪牛晓慧赵振威赵晓波陈凤阁
分析测试学报 2015年7期
李兆周,李智丽,陈秀金,李道敏,高红丽,曹 力,侯玉泽,李松彪,牛晓慧,赵振威,赵晓波,陈 慧,陈凤阁
(河南科技大学 食品与生物工程学院,河南 洛阳 471023)
乙氧酰胺苯甲酯(Ethopabate,ETP)是一种广谱抗球虫增效剂,对鸡巨型、布氏艾美耳球虫以及其他小肠球虫具有较强的杀灭作用,与其他抗球虫药物混合使用时不仅可以抑制卵囊,阻断四氢叶酸的合成,还可以降低抗药性,提高药效,因此常被制成复方制剂用于兽医临床。2010版《中国兽药典》(第一部)已收载其原料及制剂,规定其制剂的休药期为7 d。我国农业部文件《动物性食品中兽药最高残留限量》(农牧发 [2002]235号)规定:ETP在禽肝脏和肾脏中的最高残留限量为1 500 μg/kg,肌肉中的最高残留限量为 500 μg/kg[1]。
ETP在畜牧生产中长期大量应用会导致其在动物性食品中残留,从而对人类健康和环境生态具有潜在危害。建立和完善残留检测方法,定期进行监控,是控制其残留的有效途径。目前,基于各类生物性抗体的免疫分析方法需要制备结合抗原和有特异性识别作用的抗体,制备过程较为复杂,且分析过程中的稳定性较差,精密度较低。ETP的仪器检测方法主要有薄层色谱法、紫外分光光度法、高效液相色谱法[2]、高效液相色谱-串联质谱法等[3]。其中,薄层色谱法操作简单,常用作定性,但其定量的准确度和灵敏度较低。紫外分光光度法的定量误差较大,难以排除样品基质的干扰。色谱法的检出限较高,难以满足痕量兽药残留和出口贸易检测的实际需要,而且需进行步骤复杂的样品净化和富集[4]。色谱-质谱联用法的检测灵敏度高,但价格相对昂贵。由于许多动物性食品的基质较为复杂,干扰物质较多,前处理步骤比较繁琐[5]。因此,亟需开发选择性强和效果好的样品前处理和分析方法。
分子印迹技术能够获得在空间和结合位点上与某种分子完全匹配的聚合物,具有构效的预定性、识别的特异性和广泛的实用性[6]。基于该技术所制备的分子印迹聚合物(Molecular imprinting polymers,MIPs)可以特异性地识别目标化合物,且制备方法简单,稳定性好,适用于多种环境条件,使用寿命长,可多次重复使用,成本低。针对食品中的有毒有害物质,开发基于MIPs的新型分离介质,建立相应的样品处理和分析方法,成为一个方兴未艾的研究热点。
传统的MIPs制备一般采用本体聚合法,操作简便,对仪器设备条件要求低,但需要研磨、筛分和漂洗等复杂的后处理工艺,所得MIPs的形状不规则,尺寸不均匀,产率较低,且费时、费力[7]。针对本体聚合的问题,Mayes等[8]提出了悬浮聚合法用于MIPs的合成,此法可以直接生成微球形的MIPs,省去了后处理过程,大大提高了产率和吸附性能,简化了后处理工艺。在印迹聚合过程中,引发聚合条件是影响MIPs识别性能的重要因素之一,模板和单体所形成的预聚复合体越稳定,则MIPs识别性能越好[9]。悬浮聚合法多采用热分解引发剂如偶氮二异丁腈[10-11]、偶氮二异庚腈[12]等,在加热或紫外光照射条件下进行微球的制备[13],虽能成功聚合,但会影响模板-单体预聚复合体结构的稳定性,尤其是具有热敏或光敏性的模板分子。采用氧化还原引发体系可以有效解决这一问题,该引发体系能降低生成自由基的活化能,提高聚合反应的速率,进而降低反应温度,提高聚合物的识别性能[14-16]。
本文选择氧化还原引发方式,以ETP为模板分子,甲基丙烯酸为功能单体,乙腈为致孔剂,季戊四醇三丙烯酸酯为交联剂,制备并表征了具有特异性吸附能力的ETP MIPs微球。与常用的热分解引发剂相比,本文所用引发剂的引发温度较低,可大幅减少引发温度对预聚复合体的影响,提高MIPs的特异性和识别能力。所制备的微球状聚合物能最大限度地减少传统本体聚合法后处理过程中识别位点的破坏,且具有比表面积较大,分散性和均匀度较好等特点,有利于目标化合物的特异性识别[17]。所制备的MIPs有望用于食品或环境中ETP的快速、灵敏分析。
1 实验部分
1.1 试剂及仪器
ETP对照品(湖北威德利化学科技有限公司);乙酰苯胺(N-phenylacetamide,NPA)、聚乙烯醇1788、4-氨基苯甲酸甲酯(Methyl-4-aminobenzoate,MBZ)(阿拉丁试剂有限公司);乙腈、甲醇、N,N-二甲基苯胺、过氧化苯甲酰(天津市德恩化学试剂有限公司);甲基丙烯酸(MAA)、丙烯酰胺(AM)、4-乙烯基吡啶(4-VP)、季戊四醇三丙烯酸酯(美国Alfa Aesar公司)。除对照品外,所用试剂均为分析纯。
TGL-18C高速台式离心机(上海安亭科学仪器厂);1260高效液相色谱仪(美国安捷伦公司);FA1004电子分析天平(上海上平仪器有限公司);101-2A电热鼓风干燥箱(天津市泰斯特仪器有限公司);KQ3200DE超声波清洗器(昆山市超声仪器有限公司);DK-8D三孔电热恒温水槽(上海一恒科技有限公司);STARe差示扫描量热仪(瑞士梅特勒-托利多集团);VERTEX 70傅立叶变换红外光谱仪(德国布鲁克公司)。
1.2 分子印迹聚合物的制备
准确称量1.5 g聚乙烯醇1788加入100 mL蒸馏水,加热并搅拌至完全溶解,冷却至室温后转入250 mL的四口瓶中,水浴25℃,以400 r/min的转速搅拌,通氮气30 min待用。称取1 mmol ETP至25 mL烧杯,加入功能单体(甲基丙烯酸、4-乙烯基吡啶或丙烯酰胺)、交联剂(季戊四醇三丙烯酸酯)和致孔剂,超声5 min混匀;再加入引发剂过氧化苯甲酰和N,N-二甲基苯胺各0.4 mmol,超声5 min混匀。在氮气环境下,将混合溶液缓慢滴入四口瓶内,密封,以400 r/min的速度搅拌,水浴25℃引发聚合24 h。聚合反应结束后,将所得聚合物微球离心,弃去上清液,依次用蒸馏水洗涤5次,甲醇洗涤3次,乙醇洗涤3次,每次20 min。将离心的沉淀物于55℃真空干燥后,放入索氏提取器中用甲醇-乙酸混合溶液(9∶1)洗脱至无模板分子被检出,然后用甲醇洗脱除去残留溶剂,将MIPs取出,55℃干燥至恒重,以分级筛分级,获得平均粒径大小为50~70 μm的ETP分子印迹聚合物[9]。空白分子印迹聚合物(Non-molecular imprinting polymer,NIP)的合成方法除不加模板分子外,其他步骤与上述方法相同。
1.3 扫描电子显微镜表征
将MIPs微球加至载物台上,喷金处理后用场发射扫描电镜(Field emission scanning electron microscope,SEM)进行形貌观察并采集照片。
1.4 差热分析
应用差示扫描量热法(Differential scanning calorimetry,DSC)分析聚合物微球的热稳定性,起始温度25℃,空气氛围,升温速率为10℃·min-1,试样质量为9 mg。
1.5 红外光谱测定
采用KBr压片法,用傅立叶变换红外光谱仪扫描,获取MIPs的红外光谱图,根据红外光谱图中吸收峰的位置、形状、强度获得与分子结构有关的信息。
1.6 乙氧酰胺苯甲酯及其类似物分析
在进行吸附性能研究前,须建立底物的定量分析方法。参考文献报道,本研究采用高效液相色谱法对ETP及其结构类似物NPA和MBZ进行分析[2]。色谱条件:固定相为Angilent Zarbax SB C18色谱柱,250 mm×4.6 mm(i.d.);流动相为乙腈-水(35∶65)混合液;检测波长268 nm;进样量10 μL;柱温为室温;流速1 mL·min-1。
1.7 吸附性能研究
1.7.1 静态平衡吸附量 在塑料离心管中加入50 mg的MIPs和5 mL 0.5 mmol·L-1ETP的乙腈溶液,混匀,在25℃下,以200 r/min的速度振荡24 h,离心后取上清液,用乙腈稀释10倍,取1 mL注入高效液相色谱仪测定其平衡浓度,依据式(1)计算模板分子的吸附量Qw。

式中Qw为静态平衡吸附量(mmol·g-1);Cs0为底物起始浓度(mmol·L-1);Cs为吸附平衡时底物的浓度(mmol·L-1);V为底物溶液的体积(mL);m为MIPs的加入量(mg)。
依据静态吸附实验结果,计算聚合物微球的分离系数和印迹因子,根据式(2)计算底物在聚合物和溶液两相中的分离系数K。

式中,Cp为聚合物结合底物的浓度(μmol·g-1);Cs为溶液中底物的平衡浓度(mmol·L-1)。由式(3)计算聚合物微球的印迹因子IF。

其中,Kpmip为MIP的分离系数;Kpnip为NIP的分离系数。
1.7.2 等温吸附曲线 将5 mL不同浓度的ETP乙腈溶液作为吸附液,在25℃下准确称取10份50 mg MIPs,测定其对模板分子的吸附量,以吸附量对模板分子的浓度作图,绘制等温吸附曲线,将获得的数据用于式(4)的Scatchard分析[18]。根据Scatchard曲线的斜率与截距求出聚合物的平衡解离常数与最大表观吸附量。

式中,Kd为结合位点的平衡解离常数,C为模板分子的平衡浓度(mmol·L-1),Qmax为最大表观吸附量(mmol·g-1)。
1.8 选择性评价
选用印迹因子最高的MIPs进行选择性评价。将50 mg MIPs放入塑料离心管中,分别加入5.0 mL 0.5 mmol·L-1模板分子及其结构类似物的乙腈溶液(对于不溶于乙腈的物质采用甲醇溶解),置于25℃恒温气浴摇床中振荡24 h,将该混合液在20 000 g转速下离心15 min,取适量上清液用相应溶剂稀释10倍,分别测定模板分子及其类似物的平衡浓度,并计算MIPs对不同底物的Qw,K和IF[19]。
2 结果与讨论
2.1 聚合条件的优化
采用四因素三水平(L934)正交设计法优化印迹体系,根据所得聚合物的印迹因子筛选最优的反应条件和体系。在对模板分子进行印迹的过程中,致孔剂起着重要作用,既作为溶剂溶解模板分子、功能单体、交联剂和引发剂,也可以保证所制备的聚合物有合适的孔径,减少传质阻力,使模板分子及其结构类似物能够自由结合识别位点。致孔剂的理化性质与聚合物的性能间有密切关系,其极性越低,越有利于模板分子和单体之间氢键的形成。据此,本研究分别选择乙腈、氯仿和乙腈-氯仿混合溶剂(1∶1)制备印迹聚合物,根据所得聚合物的吸附性能指标优选最佳的致孔剂。此外,模板分子的加入量分别选择1,2,3 mmol;功能单体分别选择酸碱性不同的甲基丙烯酸、4-乙烯基吡啶和丙烯酰胺;根据模板分子和功能单体化学结构中的氢键作用位点,模板分子与功能单体的摩尔比分别选择1∶4,1∶6和1∶8(见表1)。
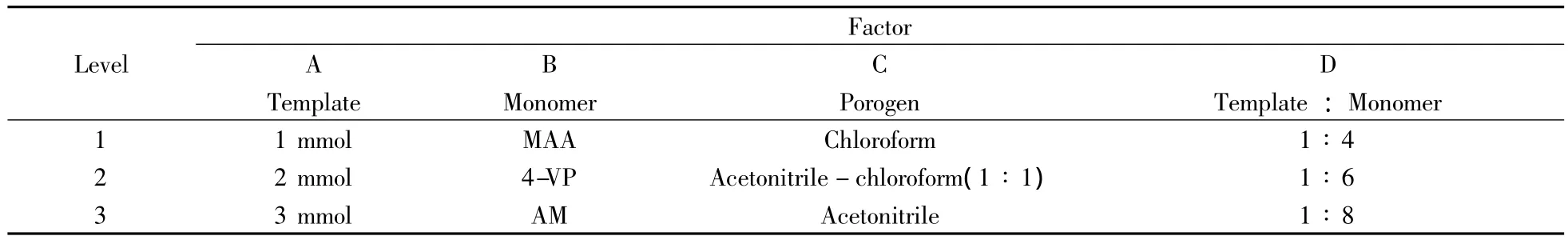
表1 正交实验因素水平表Table 1 Factors and levels for orthogonal design
不同条件下所得聚合物的IF见表2。其中K1,K2和K3为相应水平所得IF的总和,k1,k2和k3分别为K1,K2和K3的平均值,R为极差。从表可知,不同因素对所得结果影响的顺序为C>B>D>A,说明致孔剂的理化性质对模板分子和功能单体之间的非共价键相互作用的影响较大,最优的试验组合为A1B1C3D2,即:当模板分子加入量为1 mmol,功能单体为MAA,致孔剂为乙腈,模板单体摩尔比为1∶6时,所得聚合物的印迹效应最好。依此最佳组合制备MIPs,表征其各项性能指标。

表2 正交实验设计及结果Table 2 Design and results of orthogonal test

(续表2)
2.2 聚合物微球表面形貌观察
ETP印迹聚合物微球和空白聚合物微球的电镜扫描图片见图1,由图可见,聚合物微球形貌规则,表面均匀、平滑,空白聚合物微球的分布较为均匀,粒径较ETP印迹微球大(见图1A),部分ETP印迹微球表面分布有大小不等的孔洞(见图1B),可能是由于模板分子的加入抑制了聚合过程中链的延伸,致使聚合产物的分子量下降,粒径变小,分布变宽,表面孔洞增多增大。此外模板分子的加入使反应体系的极性增大,聚合物微球在反应体系中的溶解度下降而过早地沉淀出,这是ETP聚合物微球小于空白微球的另一原因[20]。

图1 ETP空白聚合物微球(A)与印迹聚合物微球(B)的电镜扫描图片Fig.1 Images of ETP NIP(A)and MIP(B)microspheres characterized by scanning electron microscope

图2 ETP MIP和NIP的红外光谱Fig.2 Infrared spectra of ETP MIP and NIP
2.3 红外光谱表征
图2为印迹聚合物和空白微球的红外图谱测定结果,在NIP微球中,由于存在MAA之间的相互作用,使得NIP的红外吸收强于MIP。在印迹过程中,模板分子的加入打破了单体间的相互作用,从而使得吸收峰明显减弱。其中,在3 000~4 000 cm-1范围内的吸收主要反映了氢键的形成,该处吸收的差异表明MIP在印迹聚合过程中形成了较强的氢键相互作用。图中1 730 cm-1为MAA分子中C‖O键的伸缩振动,在印迹过程中模板分子的—NH与MAA的C‖O基团发生了相互作用,使得C‖O键的振动减弱,吸收减少,并发生了轻度蓝移。在500~1 500 cm-1之间也出现不同程度的吸收差异,表明模板和单体的相互作用使聚合物分子结构中的多处化学键发生了振动频率和幅度改变,MIP和NIP红外图谱的差异证实了印迹效应的存在。
2.4 热稳定性分析
ETP MIP和NIP微球的DSC图谱见图3,当聚合物受热后,其吸热逐渐增多,NIP和MIP微球分别在68.6℃和83.5℃达到其吸热的峰值,随后吸热逐渐减少以至几乎没有热量吸收,此吸热过程显示上述温度为聚合物的相变温度。MIP微球由于加入了模板分子,聚合物的交联度相对较低,因此其相变温度也较低。当温度升至200℃后,聚合物逐渐呈现放热过程,推测聚合物在此温度下开始出现不同程度的分解。差热分析结果表明,聚合物的热稳定性较好,能够满足实验室和现场检测环境的需要。

图3 ETP MIP与NIP微球的DSC图谱Fig.3 DSC spectra of ETP MIP and NIP microspheres
2.5 聚合物的吸附性能
在研究吸附性能前,建立了ETP及其结构类似物NPA和MBZ的高效液相色谱定量分析方法,依据化合物的浓度(Y,mg/L)及其对应的峰面积(X)绘制标准曲线,进行定量分析[3-4],结果见表3。由表可知,所得标准曲线的线性良好,检出限(3倍信噪比)和定量下限(10倍信噪比)能够满足吸附性能分析的需要。
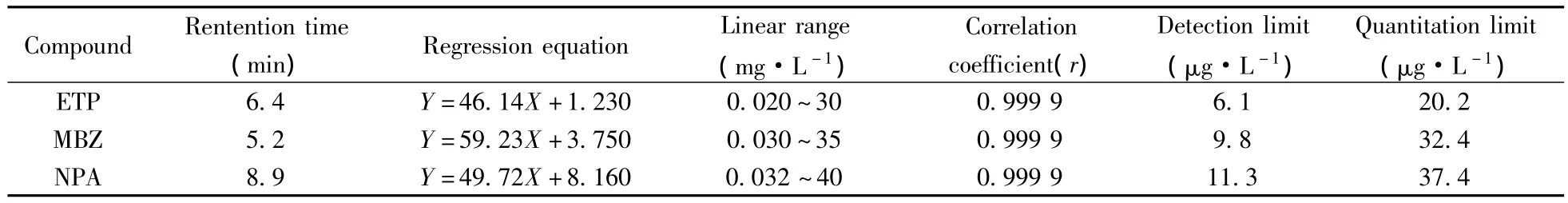
表3 ETP及其结构类似物的定量分析结果Table 3 Quantitative analysis results of ETP and structural analogues
依据各底物的定量分析方法,表征最优聚合条件下所得MIPs的吸附性能,将实验原始数据代入公式(1)~(4),聚合物的各项吸附性能参数见表4。

表4 ETP印迹聚合物吸附性能表征结果Table 4 Characterization of adsorption properties of ETP imprinted polymer
由表4可知,MIP的吸附能力显著高于NIP,其印迹因子为4.93,对ETP的吸附能力较强。进一步考察了MIP的吸附动力学曲线(见图4)。由图可知,MIP在4 h内对底物的吸附可达到稳态,NIP在2 h内即可达到稳态。这表明氧化还原法引发所得聚合物的传质阻力较低,较易达到吸附平衡,有利于聚合物对底物的快速识别。

图4 ETP印迹聚合物的吸附动力学曲线Fig.4 Kinetic adsorption curves of ETP MIPs
图5为印迹聚合物的等温吸附曲线。在25℃下,聚合物在0.7 mmol·L-1ETP的溶液中达到饱和吸附,当结合达到平衡后,单位质量MIP的吸附量随着模板分子浓度的增加而趋于饱和,且显著高于NIP的吸附量。由图可见,聚合物的吸附过程符合双分子竞争吸附的Langmuir模型。随后的Scatchard分析表明,在MIP上存在两类位点,一类是能与模板分子特异性结合的位点,对模板分子具有高度的选择性,存在于聚合物的表面和内部;另一类是非特异性的结合位点。其中MIP高亲和力位点的分离系数(KD1)和最大表观吸附量(Qmax1)分别为 0.061 μmol·L-1和 15.28 μmol·g-1;MIP低亲和力位点的分离系数(KD2)和最大表观吸附量(Qmax2)分别为 0.40 μmol·g-1和 27.12 μmol·g-1。而NIP无特异性结合位点,只有非特异结合位点,因此其达到平衡时的吸附量低于MIP。其分离系数(KD)与最大表观吸附量(Qmax)分别为0.35 μmol·L-1与 6.90 μmol·g-1。
2.6 聚合物的选择性研究
为评价聚合物的选择性,选择印迹因子最高的MIPs进行选择性评价实验,结果见表5。结果显示,以ETP为模板的MIPs对NPA和MBZ均具有一定的识别能力,印迹因子(IF)在2以上。NPA和MBZ虽然与ETP的结构非常类似,但分子结构上的极性官能团较少,这也可能是聚合物对二者识别能力稍低的原因之一,其次,底物的空间结构也会影响聚合物的识别作用。选择性评价实验表明,模板分子的结构和性质对MIPs的识别起决定性作用,这进一步印证了MIPs具有识别性能预定性的特点。
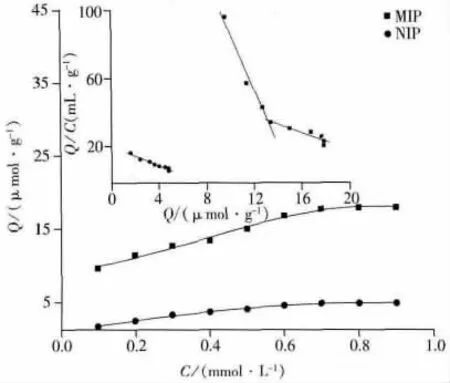
图5 ETP印迹聚合物的等温吸附曲线与Scatchard分析Fig.5 Isotherm curves and Scatchard analysis of ETP MIPs
3 结论
本文采用悬浮聚合法制备了ETP MIPs微球,结果表明,在氧化还原剂的引发下,以乙腈为致孔剂,ETP为模板分子,MAA为功能单体,季戊四醇三丙烯酸酯为交联剂,三者的摩尔比为1∶6∶20时所得聚合物的印迹效应最佳。本研究所得MIPs能够用于复杂基质中ETP的选择性识别,研究结果不仅为样品中ETP前处理和检测方法的建立奠定了基础,同时也证实了氧化还原剂引发制备MIPs微球的可行性,丰富了分子印迹技术的应用实践,为热和光不稳定模板分子MIPs的引发制备提供了参考。
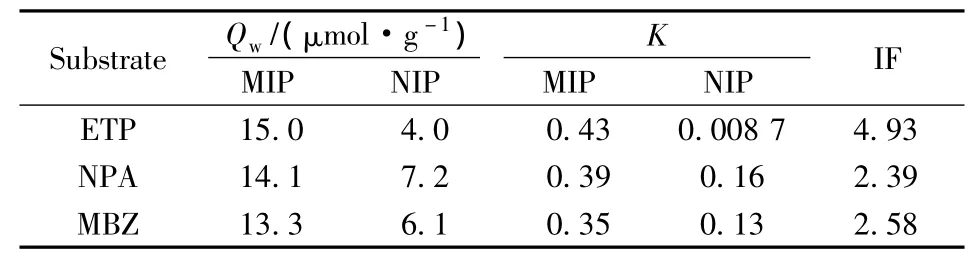
表5 ETP MIPs的选择性评价结果Table 5 The results of selectivity assessment of ETP MIPs
[1]Announcement No.235.Ministry of Agriculture of the Peoples Republic of China(235号公告.中华人民共和国农业部),2002:1-30.
[2]Granja R H M M,Nino A M M,Zucchetti R A M,Nino R E M,Salerno A G.J.AOAC Int.,2008,91(6):1483-1487.
[3]Cronly M,Behan P,Foley B,Malone E,Earley S,Gallagher M,Shearan P,Regan L.J.Pharm.Biomed.Anal.,2010,53(4):929-938.
[4]Nasr J J,Shalan S,Belal F.Food Anal.Methods,2013,6(6):1522 -1528.
[5]Chico J,Rubies A,Centrich F,Companyo R,Prat M D,Granados M.Anal.Bioanal.Chem.,2013,405(14):4777-4786.
[6]Wulff G.Microchim.Acta,2013,180(15/16):1359-1370.
[7]Lü R H,Xu L.J.Instrum.Anal.(吕瑞红,徐岚.分析测试学报),2008,27(12):1347-1350.
[8]Mayes A G,Mosbach K.Anal.Chem.,1996,68(21):3769-3774.
[9]Li Z,Qin C,Li D,Hou Y,Li S,Sun J.J.Pharm.Biomed.Anal.,2014,98:210 -220.
[10]Guo L J,Qu J R,Miao S S,Geng H R,Yang H.J.Sep.Sci.,2013,36(24):3911 -3917.
[11]Balamurugan K,Gokulakrishnan K,Prakasam T.Saudi Pharm.J.,2012,20(1):53 -61.
[12]Krstulja A,De Schutter C,Favetta P,Manesiotis P,Agrofoglio L A.J.Chromatogr.A,2014,1365:12-18.
[13]Lai J P,Sun H,Chen F,Fan L,Liu G L,Lin D S,Yang Z.J.Instrum.Anal.(赖家平,孙慧,陈芳,樊莉,刘桂伶,林东升,杨洲.分析测试学报),2012,31(9):1161-1169.
[14]Ginzburg-Turgeman R,Mandler D.Phys.Chem.Chem.Phys.,2010,12(36):11041 -11050.
[15]Nussbaum D A,Gailloud P,Murphy K.J.Vasc.Interv.Radiol.,2004,15:121 -126.
[16]Milner R.J.Biomed.Mater.Res.B,2004,68B(2):180-185.
[17]Khorrami A R,Edrisi M.Sep.Sci.Technol.,2010,45(3):404 -412.
[18]Yi L X,Fang R,Chen G H.J.Chromatogr.Sci.,2013,51(7):608 -618.
[19]Cheong W J,Yang S H,Ali F.J.Sep.Sci.,2013,36(3):609 -628.
[20]Zhang X,Fu Z F,Shi Y,Dong Z J.J.Beijing Univ.Chem.Technol.:Nat.Sci.Ed.(张笑,付志峰,石艳,董志佼.北京化工大学学报:自然科学版),2011,38(1):72-76.
猜你喜欢
陶瓷研究(2022年3期)2022-08-19
云南化工(2021年6期)2021-12-21
云南画报(2021年10期)2021-11-24
潍坊学院学报(2020年6期)2020-11-22
科学(2020年2期)2020-08-24
小学生优秀作文(高年级)(2018年4期)2018-09-11
大连工业大学学报(2015年4期)2015-12-11
生物技术通报(2015年1期)2015-04-10
中国当代医药(2015年29期)2015-03-01
中国摄影(2014年12期)2015-01-27
