
甲醛在CeO2(110)表面的吸附
2016-09-01 07:21姚小丹韩姣囡金丽芳刘晓娜蒋仕宇滕波涛
广州化工 2016年5期
姚小丹,韩姣囡,金丽芳,刘晓娜,蒋仕宇,滕波涛
(浙江师范大学化学与生命科学学院,浙江 金华 321004)
甲醛在CeO2(110)表面的吸附
姚小丹,韩姣囡,金丽芳,刘晓娜,蒋仕宇,滕波涛
(浙江师范大学化学与生命科学学院,浙江金华321004)
利用密度泛函理论计算了甲醛在洁净CeO2(110)表面的吸附行为,发现甲醛在表面存在化学与物理吸附。甲醛化学吸附时,甲醛的碳、氧原子分别与表面氧、铈原子发生作用,生成CH2O2,平面结构变为四面体构型;吸附能随覆盖度的减小而增大;电子结构分析表明,其最高占据轨道nO的电子进入表面铈原子的空轨道,形成Ce-Os化学键;而表面氧原子的电子填充到甲醛的C-Of反键轨道,形成新的C-Os键,甲醛的C-Of键长伸长。而物理吸附的甲醛,平面结构不变,吸附能较小,电子结构无明显变化。
密度泛函理论;CeO2(110);吸附能;态密度
目前,甲醛已成为最主要、最严重的室内污染源之一,可严重危害人体健康,如何消除室内甲醛污染引起人们的广泛重视[1-2]。以氧化铈负载金属的多相催化氧化技术可将室内空气中的甲醛氧化为二氧化碳与水,成为有效消除室内甲醛污染的重要方法之一[3-11]。
实验研究表明,不同形貌的氧化铈纳米粒子暴露不同指数的终端表面,其中氧化铈纳米颗粒暴露(111)表面;纳米棒暴露(111)与(110)表面;纳米立方体通常暴露(100)表面。不同指数终端的表面其微观结构不同,导致其相应的催化氧化活性不同。因此,有必要对甲醛在不同终端的纳米粒子表面的吸附与反应行为进行深入的研究。
Zhou等[12]利用TPD、SXPS及NEXAFS研究了甲醛在CeO2(111)表面的吸附行为,发现甲醛在洁净的(111)表面吸附形成CH2O2物种;当温度升高时,表面吸附的CH2O2物种发生脱附生成甲醛分子。蒋等[13-14]就甲醛在CeO2表面吸附的理论研究进行了有益的尝试,发现甲醛在CeO2(111)表面形成了吸附的CH2O2物种,同时对形成相应物种的电子结构进行了解释。但目前的理论研究对甲醛在CeO2(110)表面的吸附与反应行为鲜有报道。
本章利用密度泛函理论,对甲醛在洁净CeO2(110)表面的吸附行为进行了系统研究,讨论了甲醛覆盖度对吸附行为的影响,对不同吸附位的几何结构、吸附能及电子结构进行了深入分析,为进一步探讨甲醛在不同形貌的CeO2纳米粒子上的吸附与氧化反应行为奠定了基础。
1 模型与计算方法
本文计算工作采用基于密度泛函理论的VASP[15]程序包。电子交换关联势采用GGA-PW91计算,离子实采用投影缀加平面波赝势(PAW)[16]描述,Kohn-Sham单电子态采用平面波基组展开,其截止能设置为400 eV。理论计算的能量与结构优化标准设定为:(1)自洽场能量收敛标准为1.0×10-4eV;(2)最大力设置为0.03 eV·Å-1,优化收敛的能量小于1.0×10-3eV。
为系统研究不同覆盖度对甲醛在CeO2(110)吸附行为的影响,本文选取p(1×1)、(1×2)及(1×3)的CeO2(110)周期性四层原子平板模型,其中下面的两层原子固定,上面两层原子弛豫,如图1所示。CH2O自由分子在15 Å×15 Å×15 Å的晶胞中进行结构优化及能量计算。优化得到的甲醛结构参数如下:dC-O=1.212 Å;dC-H=1.117 Å;∠HCO=124.34°;∠HCH=111.33°
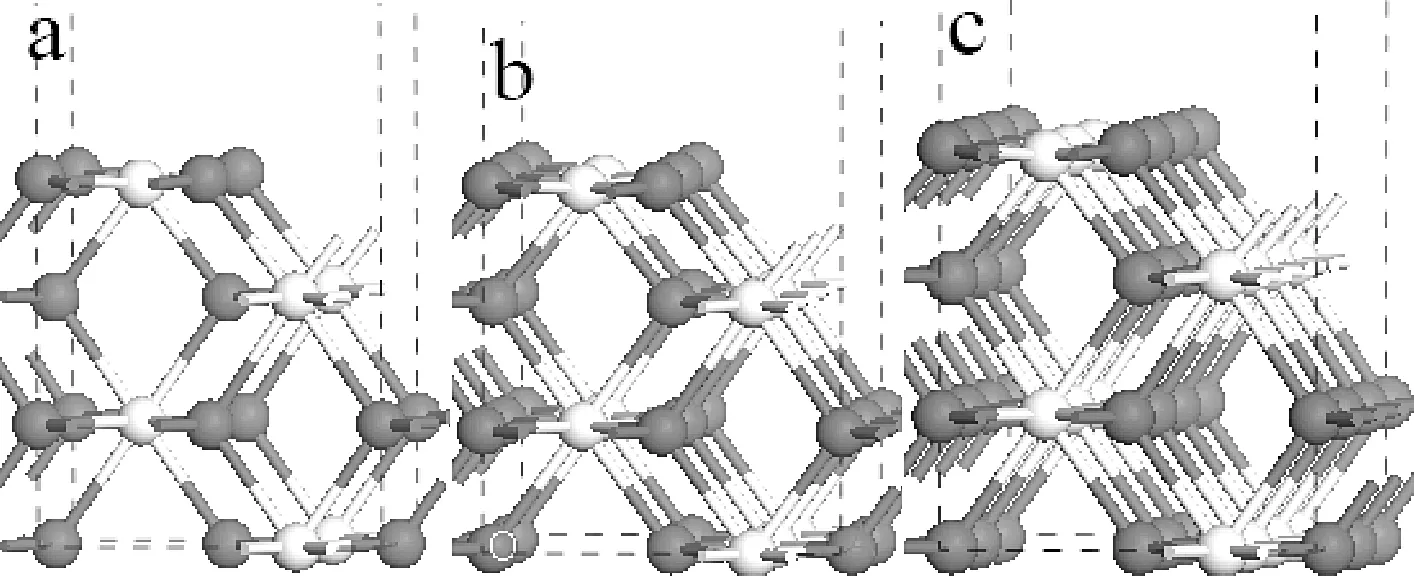
图1 CeO2(110)-p(1×1)、 (1×2)与(1×3)超晶胞模型
CH2O的覆盖度的定义为:θ= nCH2O/nOs,其中nCH2O表示吸附的CH2O的数目,nOs表示氧化铈表面氧原子的数目。故甲醛在CeO2(110)p(1×1)、p(1×2)与p(1×3)模型对应的覆盖度分别为0.50,0.25,0.17 ML (Monolayer)。
不同周期性平板模型的布里渊区积分采用Monkhorst-Pack[17]方法k点取样。对CeO2(110)的p(1×1)、p(1×2)与p(1×3)模型选取的k点分别为4×6×1、4×3×1与4×2×1。为消除相邻层间甲醛分子与表面的影响并保证表面原子层有足够的自由距离,周期性平板模型真空层厚度取12 Å。通过收敛性的测试,以上设定足以保证计算的精确度。
甲醛在CeO2(110)表面的吸附能定义为:
Ead=E(CH2O/surface)-[E(CH2O)+E(surface)]
式中,E(CH2O/surface)是CeO2表面吸附甲醛后的总能量,E(CH2O)与E(surface)是吸附前甲醛自由分子与洁净CeO2(110)表面的能量。根据该定义,Ead为负值表明是放热吸附过程,Ead为正值则是吸热过程。
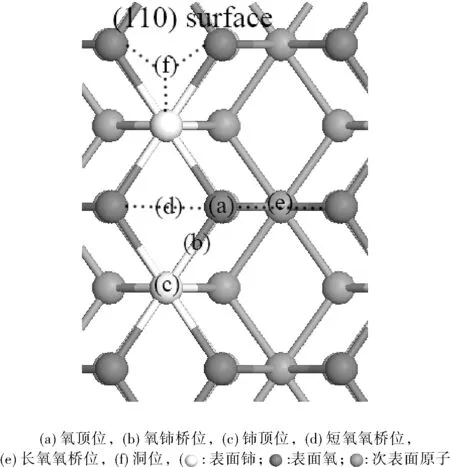
图2 CeO2(110)表面吸附位
如图2所示,甲醛在CeO2(110)表面可能的吸附位有:(a)氧原子的顶位,记为O-top;(b)氧、铈原子间桥位,记为O-Ce-bridge;(c)铈原子的顶位,记为Ce-top;(d)两较近氧原子间的桥位,O-O-s-bridge;(e)两较远氧原子形成的桥位,记为O-O-l-bridge;(f)两较近氧原子与铈原子组成的洞位,记为hollow位。甲醛的碳、氢原子可以吸附在CeO2表面的氧原子上,相应碳、氧原子可以吸附在表面铈原子上。由模型表面不同的吸附位与甲醛不同吸附原子组合获取甲醛吸附的初始构型。
2 结果与讨论
2.1吸附构型与吸附能
与甲醛在CeO2(111)表面的吸附相似,甲醛在CeO2(110)表面吸附亦分为两种情况:(a)甲醛的氧原子与碳原子吸附在表面的铈原子与氧原子上的化学吸附;(b)甲醛的氧原子吸附在表面铈原子上的物理吸附。具体优化构型如图3所示,相应几何结构参数与吸附能见表1。
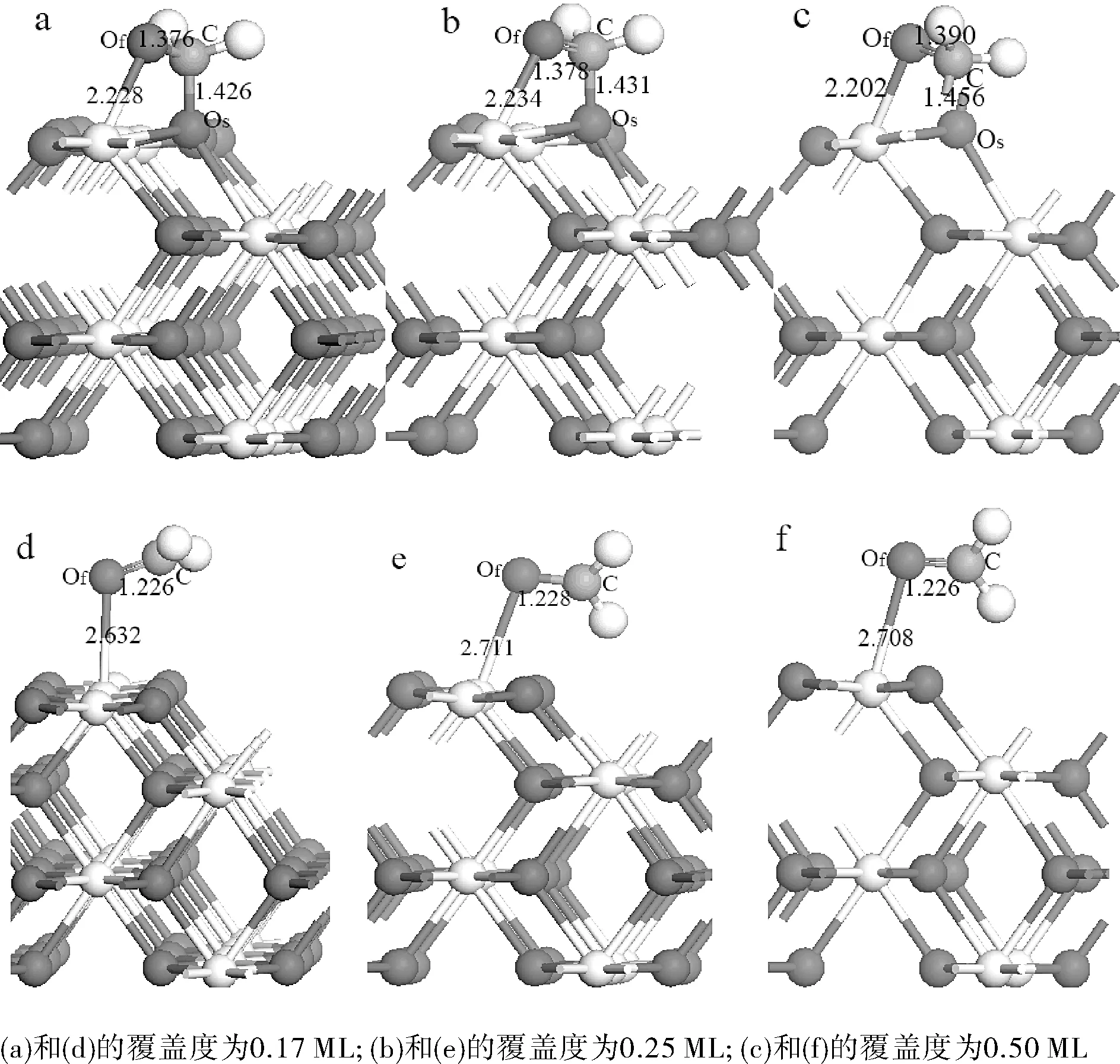
图3 甲醛在CeO2(110)表面吸附构型
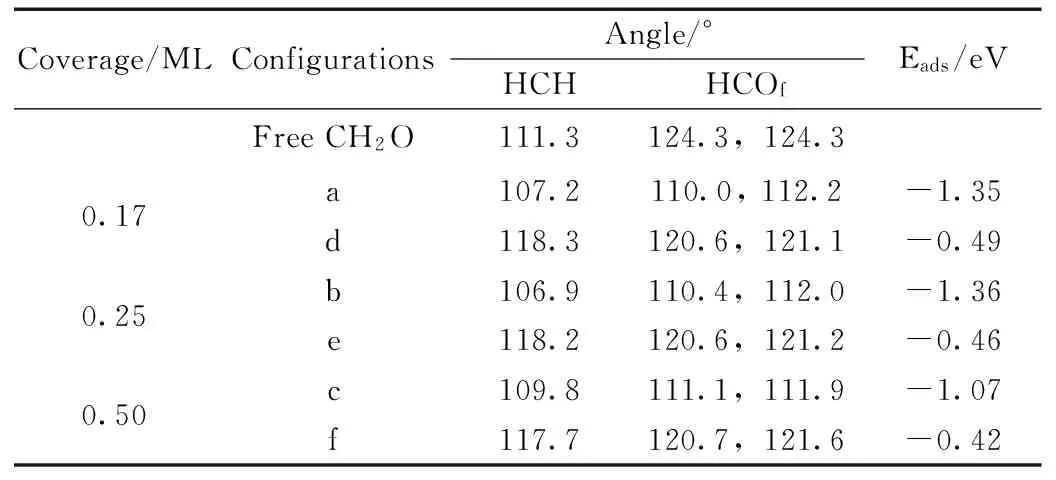
表1 CH2O自由分子及其在CeO2(110)表面吸附的几何构型参数与吸附能
2.1.1低覆盖度
如图3(a)所示,当甲醛在CeO2(110)-p(1×3)表面(0.17 ML)吸附时,甲醛的碳原子与氧原子分别吸附在CeO2(110)表面的氧与铈原子上,同时甲醛分子的构型会发生明显的变化:甲醛的两个氢原子往上翻转,C原子呈sp3杂化结构;而当甲醛的氧原子吸附在CeO2(110)表面的铈原子上时,如图3(d)所示,平面结构的甲醛没有发生明显的构型改变,仍近似为平面构型,只是发生一定程度的翻转。
从被吸附甲醛的键长来看,相对自由甲醛分子,无论是发生物理吸附还是化学吸附,被吸附甲醛的C-H键长均不发生明显的改变,物理吸附构型的C-Of键长增长约0.02 Å,为1.228 Å;化学吸附的甲醛其C-Of键长增大为1.378 Å。HCH与HCOf键角也有所变化:物理吸附的甲醛其HCH与HCOf键角分别约为118°与121°,而化学吸附构型的相应键角则分别为107°与111°。
从吸附能来看,当甲醛物理吸附在CeO2(110)表面时,吸附能为-0.49 eV;而当其发生化学吸附时,其吸附能达-1.35 eV,为强放热吸附,并较其在CeO2(111)表面吸附能约高0.5 eV,这是由于CeO2(110)具有开放的表面结构, O、Ce原子同时位于表面,可以更好地与甲醛分子的C、O原子作用,导致其吸附能增强。
当甲醛覆盖度增加至0.25 ML时,其吸附构型与甲醛在CeO2(110)的p(1×3)模型表面吸附结构基本一致,相应吸附键长、键角及吸附能亦基本一致,如表1所示,说明吸附的甲醛分子之间的相互作用可忽略不计。
2.1.2高覆盖度
当甲醛覆盖度进一步增加至0.5 ML时,即甲醛吸附在CeO2(110)的p(1×1)超晶胞表面模型,相应优化构型见图3(c、f)。由图3(c)可知,被吸附甲醛的构型与高覆盖度构型基本一致,即平面结构的甲醛自由分子变为四面体构型。对比几何构型参数可知,甲醛的C-H键长没有发生明显改变,但C-Of键长却增大约0.18 Å;同时由于甲醛氢原子往上翻转,导致HCH与HCOf键角都变小,分别约为110°与111°。从图3(f)可以看出,与自由甲醛分子相比,物理吸附构型的甲醛其键长(C-H与C-Of键长)均没有明显变化,HCOf键角变化也不大(为121°),但HCH键角却增大为118°(约增大6%)。由表1中吸附能数据可知,甲醛在CeO2(110)的p(1×1)表面的吸附均为放热过程,物理吸附的甲醛吸附能为-0.42 eV;而化学吸附的甲醛吸附能为-1.07 eV,均弱于低覆盖度的情况,这是由于高覆盖度时,甲醛分子间相互排斥作用增强,导致其与表面作用力减弱。
从以上分析可以看出,对不同的覆盖度下物理吸附或化学吸附的甲醛构型相近。分析吸附的甲醛与表面间的距离可知,当发生化学吸附时,甲醛与表面之间会生成新的Of-Ce键与C-Os键;而物理吸附的甲醛与表面间的相互作用则相对较弱。从表1可知,物理吸附构型的甲醛其氧原子与表面铈原子之间的距离为2.7 Å,大于Ce-O间成键距离,而化学吸附构型的甲醛氧原子与表面的铈原子间的Of-Ce键长以及甲醛的碳原子与表面氧原子间的C-Os键长均在其成键范围(分别为2.2 Å与1.5 Å)之内,即甲醛与表面之间有Of-Ce键及C-Os键生成。
2.2电子结构
为从电子角度对甲醛与CeO2(110)表面的相互作用进行分析,本文系统计算了不同覆盖度下甲醛在CeO2(110)表面的总态密度及局域态密度,结果如图4所示。
从图4(a、c、e、g)CeO2(110)、CH2O/CeO2(110)体系的电子态密度(DOS)图可以看出,吸附甲醛后的CeO2(110)表面电子态密度图仍保持洁净CeO2(110)表面的特征峰:(1)在-12 eV以下的宽峰为Ce的5p电子;(2)在-4.5~0 eV的宽峰为氧的2p价带;(3)费米能级以上为铈的4f非占据窄峰,即为体系的导带。另一方面,吸附甲醛后的CeO2(110)表面DOS峰略微向低能量方向移动;同时由于表面吸附甲醛,在-10 eV附近及-6~-5 eV处出现甲醛的特征DOS峰。
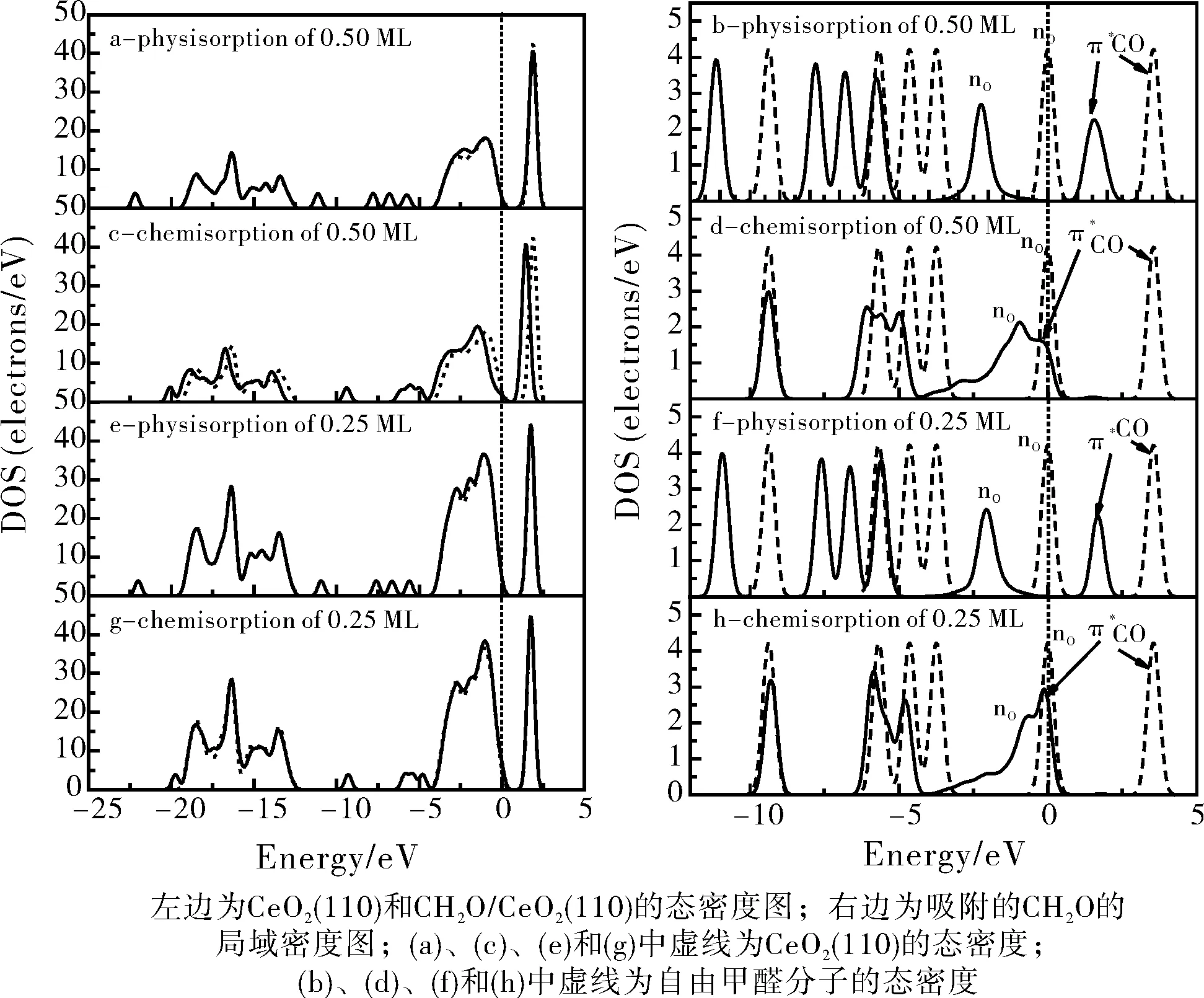
图4 甲醛在CeO2(110)表面吸附的DOS图
如图4(b)与(f)所示,当甲醛物理吸附时,其电子峰位置往低能量方向偏移,说明甲醛在CeO2(110)表面发生物理吸附后体系的总能量降低,同时甲醛部分电子向表面转移;另一方面,其四个电子峰分别与自由甲醛分子的σ’CH、σCO、πCO与nO峰一一对应,物理吸附的甲醛各轨道变化不大,甲醛与CeO2(110)表面作用较弱,与其较低的吸附能计算结果相一致。

3 结 论
本文利用密度泛函理论研究了甲醛在CeO2(110)表面的吸附情况。结果表明:甲醛在CeO2(110)表面的吸附存化学吸附与物理吸附;物理吸附的甲醛仍近似为平面结构,其吸附能较小,其电子结构与自由甲醛分子相似;化学吸附的甲醛中碳、氧原子分别与表面氧、铈原子作用,形成CH2O2物种;电子结构分析发现,其最高占据轨道nO的电子进入表面铈原子的空轨道,形成Ce-Os化学键;而表面氧原子的电子填充到甲醛的C-Of反键轨道,形成新的C-Os键,同时甲醛的C-Of键长伸长;甲醛在CeO2(110)表面的吸附能随着覆盖度的增大而减小,当覆盖度降低为0.25 ML时,吸附甲醛分子间的相互作用可以忽略。
[1]何运兵,纪红兵,王乐夫.室内甲醛催化氧化脱除的研究进展[J].化工进展,2007,26(8): 1104-1109.
[2]岳伟,潘小川.室内空气污染物及其健康效应研究[J].环境与健康杂志,2005,22(2): 150-152.
[3]Jia M L, Shen Y N, Li C Y, et al. Effect of supports on the gold catalyst activity for catalytic combustion of CO and HCHO [J].Catal. Lett., 2005,99(3-4): 235-239.
[4]Tang X F, Chen J L, Li Y G, et al. Complete oxidation of formaldehyde over Ag/MnOx-CeO2catalysts [J]. Chem. Eng. J., 2006, l18(1-2): 119-125.
[5]Zhang C B, He H, Tanaka K. Catalytic performance and mechanism of a Pt/TiO2catalyst for the oxidation of formaldehyde at room temperature [J]. Appl. Catal. B, 2006, 65(1-2):37-43.
[6]Yang X Z, Shen Y N, Yuan Z F, et al. Ferric ions doped 5A molecular sieves for the oxidation of HCHO with low concentration in the air at moderate temperatures [J]. J. Mol.Catal. A-Chem., 2005, 237(1-2): 224-231.
[7]Imamura S, Uchihori D, UtaniK.Oxidative decomposition of formaldehyde on silver-cerium composite oxide catalyst[J]. Catal. Lett., 1994, 24(3-4): 377-384.
[8]Saleh J M, Hussian S M. Adsorption, desorption and surface decomposition of formaldehyde and acetaldehyde on metal films nickel, palladium and aluminium[J]. J. Chem. Soc. Faraday Trans., 1986, 82(1): 2221-2234.
[9]Peng J X, Wang S D.Performance and characterization of supported metal catalysts for complete oxidation of formaldehyde at low temperatures[J]. Appl. Catal. B-Environ., 2007, 73(3-4): 282-291.
[10]Shen Y N, Yang X Z, Wang Y Z, et al. The states of gold species in CeO2supported gold catalyst for formaldehyde oxidation [J]. Appl. Catal. B-Environ., 2008, 79(2): 142-148.
[11]Li C Y, Shen Y N, Jia M L, et al.Catalytic combustion of formaldehyde on gold/iron-oxide catalysts [J]. Catal. Commun., 2008, 9(3): 355-361.
[12]Zhou J, Mullins D R.Adsorption and reaction of formaldehyde on thin-film cerium oxide [J]. Surf. Sci., 2006, 600(7): 1540-1546.
[13]蒋仕宇,滕波涛,鲁继青,等.甲醛在CeO2(111)表面吸附的密度泛函理论研究[J].物理化学学报, 2008, 24(11):2025-2031.
[14]蒋仕宇. 铈基催化剂上甲醛催化氧化的密度泛函理论研究[D]. 金华:浙江师范大学化学与生命科学学院,2009
[15](a) Kresse G,Hafner J.Ab initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium[J]. Phys. Rev. B., 1994,49(20): 14251-14269;(b)Kresse G, Furthmiiller J.Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set [J]. Comput. Mater. Sci., 1996, 6(1): 15-50.
[16]Kresse G, Joubert D. From ultrasoftpseudopotentials to the projector augmented-wave method[J]. Phys. Rev. B 1999,59: 1758-1775.
[17]MonkhorstH J, Pack J D.Special points for Brillouin-zone integrations[J]. Phys. Rev. B, 1976, 13(12): 5188-5192.
Adsorption of Formaldehyde on CeO2(110) Surface
YAOXiao-dan,HANJiao-nan,JINLi-fang,LIUXiao-na,JIANGShi-yu,TENGBo-tao
(College of Chemistry and Life Sciences, Zhejiang Normal University, Zhejiang Jinhua 321004, China)
density functional theory; CeO2(110); adsorption energy; density of states
姚小丹(1992- ),女,研究生,主要从事模型催化剂的理论研究。
O643.36
A
1001-9677(2016)05-0134-04
猜你喜欢
科学技术创新(2022年30期)2022-10-21
农业与技术(2021年23期)2021-12-14
椰城(2021年12期)2021-12-10
黑龙江水利科技(2020年8期)2021-01-21
少儿科学周刊·少年版(2021年22期)2021-01-17
原子与分子物理学报(2020年5期)2020-03-17
青岛大学学报(工程技术版)(2019年2期)2019-09-10
物理学报(2017年21期)2017-11-10
腐蚀与防护(2016年7期)2016-09-14
枣庄学院学报(2015年5期)2016-01-09
