
ATP1A3基因突变致儿童交替性偏瘫2例报告并文献复习
2017-04-06 12:37马建南
临床儿科杂志 2017年2期
张 婷 马建南 肖 农
重庆医科大学附属儿童医院 儿童发育疾病研究教育部重点实验室 儿童发育重大疾病国家国际科技合作基地 儿科学重庆市重点实验室(重庆 400014)
ATP1A3基因突变致儿童交替性偏瘫2例报告并文献复习
张 婷 马建南 肖 农
重庆医科大学附属儿童医院 儿童发育疾病研究教育部重点实验室 儿童发育重大疾病国家国际科技合作基地 儿科学重庆市重点实验室(重庆 400014)
目的探讨儿童交替性偏瘫的临床表现、基因诊断及治疗方法。方法回顾性分析2例交替性偏瘫患儿的临床资料,并进行相关文献复习。结果2例患儿均为女性,分别于4月龄、6月龄起病,1例的首发症状为交替性偏瘫,另1例的首发症状为惊厥,并于病程第2年出现交替性偏瘫。ATP1A3基因测序显示,2例患儿分别存在c.2401G>A (p.D801N)和c.2731G>C (p.A911P)杂合错义突变,后者在人类基因突变数据库(HGMD)专业版中尚未见报道。结论对临床诊断交替性偏瘫的患儿,建议行ATP1A3基因筛查,有助于确诊及遗传咨询。
交替性偏瘫; ATP1A3基因; 儿童
儿童交替性偏瘫(alternating hemiplegia of childhood,AHC)是一种罕见的神经系统发作性疾病,于1971年由Verret等[1]首先报道。AHC多数在生后18个月内起病,主要表现为反复发作的交替性偏瘫、发作性的眼球运动异常、肌张力不全,少数患儿可合并癫痫发作,绝大多数患儿智力运动发育落后于同龄儿童[2]。既往该病诊断主要依靠临床表现,直到2012年,Heinzen和Rosewich等[3,4]发现该病的主要致病基因为ATP1A3,且证实了多数为新发突变,使该病可以从分子遗传学角度得到确诊。现报告2例经基因检测明确诊断的AHC。
1 临床资料
例1,女,1岁1个月。4月龄起病,首发症状为发作性单侧肢体无明显活动,每次发作持续3~4 d后自行缓解,单侧肢体无力在清醒时出现,睡眠时好转,每月发作1~2次,双侧肢体可交替出现无力表现。病程中,有阵发性双目向患侧斜视,伴颈部偏向同侧,无意识丧失,有阵发性一侧肢体僵硬。患儿于6月龄时,在当地医院就诊,脑电图示睡眠期右侧额区可疑尖/尖慢波,诊断癫痫,予以奥卡西平口服治疗,患儿症状无好转。患儿围生期无缺氧缺血病史,有语言及大运动发育落后表现,就诊时尚不能说话,不能独站。家族史无类似表现者,否认有癫痫家族史。患儿入院时体格检查示神清、反应可,无特殊面容,无特殊皮疹,头围46 cm,双侧瞳孔等大等圆,光反射灵敏,四肢活动可,活动对称,肌张力正常。入院后实验室检查示肝肾功能、电解质、头颅磁共振成像(MRI)平扫及动静脉血管成像、视频脑电图均无异常。
经医院伦理委员会审核,在患儿父母签署知情同意书后,患儿及其父母均采集2 mL全血,行Sanger双脱氧链终止法检测ATP1A3基因。结果显示,患儿ATP1A3基因存在杂合突变:c.2401G>A (p.D801N),该突变已有报道[4]。父母均未检测到此突变,考虑为自发突变。见图1。
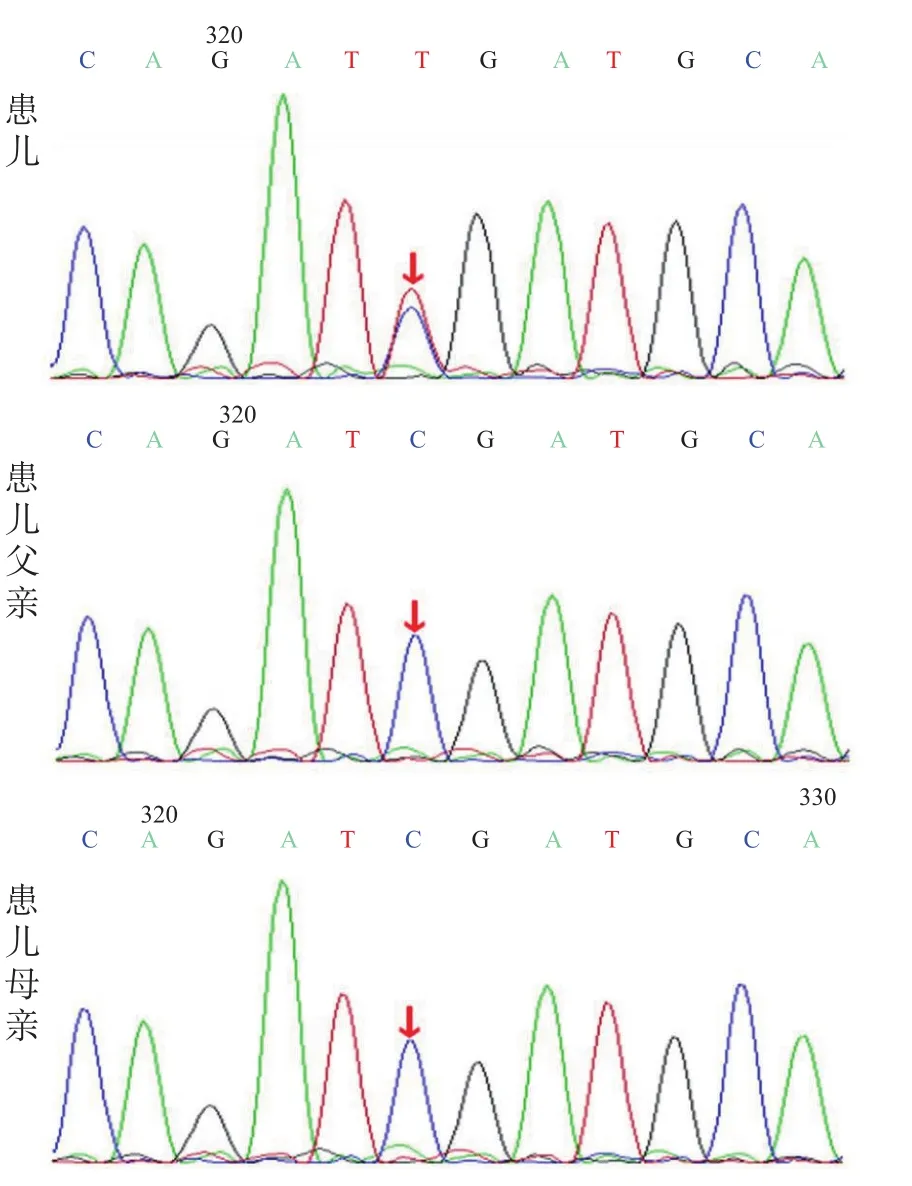
图1 例1及父母ATP1A3基因测序图
确诊后予氟桂利嗪5 mg/d治疗,随访半年,患儿偏瘫发作频率减少,共发生2次,每次持续时间缩短为1~2 d,阵发性眼球偏斜频率亦有减少。
例2,女,现9岁。6月龄时起病,首发症状为癫痫发作,表现为全身强直-阵挛发作,2、3次/年,病程第2年出现发作性交替性偏侧肢体无力,偶伴运动性失语,每次发作持续时间为数分钟至数天不等,发作频率为3次/d至2次/月,睡眠或休息后发作可缓解,偶有阵发性一侧肢体僵硬,无意识丧失。发作期间可伴有阵发性头晕、非喷射性呕吐,偶有视物重影。病初诊断为癫痫,予以丙戊酸钠治疗,癫痫发作停止,但仍有发作性单侧无力表现;家属自行停药后再予妥泰治疗,患儿发作性肢体无力频率较用药前减半,无癫痫发作。患儿围生期无异常;智力及运动发育落后,1岁7个月才能独走。否认家族中有类似表现者,否认有癫痫家族史。患儿的舅舅及伯父早夭,但原因不详。入院体格检查示发育迟滞,神志清楚,无特殊面容,表情稍幼稚,理解力、计算力差,时间及空间定向力正常;双侧瞳孔等大等圆,直径0.4 cm,对光反射灵敏;右上肢肌力Ⅱ级,右下肢肌力Ⅱ~Ⅲ级,左侧上下肢肌力Ⅴ级,四肢肌张力正常;双侧膝反射++,双侧巴氏征阴性,脑膜刺激征阴性。入院后头颅MRI检查未见明显异常,磁共振血管造影及磁共振静脉成像示左侧横窦未见显影,左侧乙状窦显影浅淡,较对侧纤细,余颅内大血管及其大分支未见明显异常。视频脑电图清醒期3~5 Hz δθ混合活动阵发,清醒期短(约3 min),不除外思睡期因素影响。肝肾功电解质均无异常。
经医院伦理委员会审核,患儿父母签署知情同意书后,患儿及其父母均抽2 mL全血,行Sanger双脱氧链终止法检测ATP1A3基因。结果显示,患儿ATP1A3基因存在杂合突变:c.2731G>C (p.A911P),见图2。该突变为错义突变,虽然在人类基因突变数据库(Human Gene Mutation Database,HGMD)专业版数据库中尚未见报道,但由于其不属于多态性位点,在人群中发生频率极低,且结合患儿临床有交替性偏瘫的表现,故考虑为致病突变。父母均未检测到此突变,考虑为新发突变。
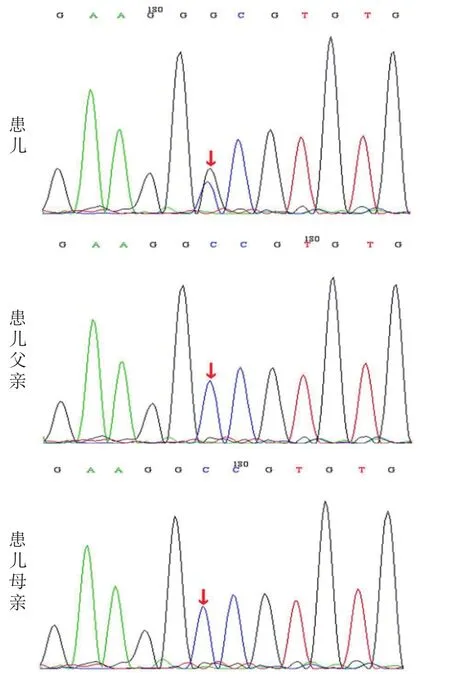
图2 例2及父母的ATP1A3基因测序图
诊断后予以氟桂利嗪10 mg/d治疗及妥泰抗癫痫治疗,随访1年,患儿偏瘫发作共2次。
2 讨论
AHC是一种罕见的神经系统发作性疾病,其患病率大约为1/100 0000[5],但由于其发作性症状的多样性容易被误诊,所以其患病率可能被低估了。在AHC的致病基因未明确前,本病仅依靠临床诊断,比较公认的标准是1993年Bourgeois等[6]提出的诊断标准:①起病年龄在18个月以内;②反复发作的偏瘫,双侧肢体交替出现;③发作性四肢瘫或全瘫;④症状在睡眠后缓解且醒后可再次发生;⑤其他发作性症状,包括发作性肌张力障碍、眼球运动异常和自主神经症状,以上症状可以与偏瘫发作同时或单独发生;⑥智力低下或其他神经系统异常,如共济失调、舞蹈手足徐动。
为了使临床医师更好地掌握本病,Mikati等[7]将AHC的病程分为三个阶段。第一阶段一般在出生后数月内到1岁左右,以异常眼球运动及肌张力不全发作为主要表现,此期偏瘫发作往往不常见。第二阶段持续约1~5年,此期出现偏瘫发作的典型表现,若既往已有偏瘫发作者则发作更为频繁,同时伴有智力运动发育的落后,发作间期常有发作性肌张力不全及舞蹈手足徐动症。第三阶段往往偏瘫发作明显减轻,但遗留不可逆认知损害。
然而,临床上本病仍然容易漏诊或误诊,一方面因为临床表现复杂多样,另一方面并非所有病例均符合典型的三阶段特点。国内总结78例交替性偏瘫患儿的首发症状,表现为眼球运动异常的38例(48.7%),肌张力不全13例(16.7%),眼球运动异常合并肌张力不全5例(6.4%),偏瘫17例(21.8%),四肢瘫3例(3.8%),癫痫发作2例(2.6%)。本研究的例1患儿首发症状为交替性偏瘫,因此在病程的第一年就得到了确诊;例2患儿的首发症状为惊厥,虽然在病程的第2年即出现了发作性交替性偏瘫等典型症状,但在长达8年的时间内均只被诊断为癫痫,而忽视了交替性偏瘫。另外,当患儿的首发症状为异常眼球活动及肌张力不全发作时,也常常被误诊为癫痫这一最常见的儿童发作性疾病。提示临床医师在面对发作性疾病的患儿时,需仔细询问病史,特别要注意发作的表现及其变化。
AHC虽多数为散发病例,但既往国内外均有AHC家系的个案报道[8,9],提示遗传因素可能是其致病原因。随着分子生物学技术的发展,2012年Heinzen和Rosewich等[3,4]证实了ATP1A3基因是AHC的致病基因。ATP1A3基因位于19q13.2,包含23个外显子,是编码Na+/K+-ATP酶α3亚单位的基因,它主要在神经元细胞表达。Na+/K+-ATP酶一种跨膜蛋白,主要功能是完成细胞内外的钠、钾离子的交换,从而维持细胞内外的钠、钾浓度梯度。它由1个α亚单位和1个β亚单位组成,其中α亚单位共有4种异构体(α1~α4),分别由不同的基因编码。在ATP1A3基因突变被报道是导致AHC的主要病因之前,该基因突变最早在速发型肌张力不全-帕金森综合征(rapidonset dystonia-parkinsonism,RDP)的家系中被报道[10]。RDP多为成年期急性起病,其主要表现是肌张力不全和帕金森综合征样症状,常常由一些物理因素或情绪刺激诱发起病。AHC和RDP虽然主要致病基因均为ATP1A3,但两种疾病的基因突变位点分布形式和发生碱基改变不同[11]。对ATP1A3基因突变体的体外功能研究证明,AHC相关ATP1A3基因突变引起了蛋白活性降低,但没有影响蛋白表达量;而RDP相关ATPlA3基因突变不仅引起蛋白表达下降,同时引起了蛋白活性降低[3]。
迄今,国外研究报道的AHC病例中ATPlA3基因突变率为78%~100%[3,4,13],绝大多数突变为新生突变,其中,最常见的两种突变为D801N9(C.2401G>A)和E815K(C.2443G>A)。国内报道筛查78例AHC患儿,ATP1A3基因突变率为91%,其中3例为家系病例,95.5%被证实为新生突变,共发现27种杂合错义突变,最常见的突变除了D 801 N(28.2%)和E 815 K(16.9%),还有G 947 R(c.2839 G>A)(9.7%)和G947R(c.2839G>C)(7.1%)[14]。本研究的例1患儿的突变为D801N(c.2401G>A),为热点突变,与国内外文献报道一致;例2患儿的突变为A911P(c.2731G>C),目前国内外尚无文献报道。值得一提的是,国内外报道的AHC患者ATP1A3基因的新发突变率并非都是100%,这提示可能除了新发点突变以外还可能存在其他的突变方式(如:大片段缺失或重复)或存在其他基因突变,今后的遗传学研究除了应关注AHC的基因型和表型之间的联系,同时应该寻找新的突变方式或致病基因。另外,国内外均有报道起病晚于18个月的ATP1A3基因突变检测为阳性的不典型AHC[3,4,14],提示对于少数起病年龄晚于18个月的不典型病例,若经检测其存在ATP1A3基因突变,在满足AHC其他诊断标准时,也可以诊断AHC。
目前,AHC尚无特异性的治疗方法。在发作期,由于患儿睡眠时无肢体瘫痪表现,可通过给予镇静药物(如水合氯醛、安定或咪达唑仑)来增加睡眠时间,从而缩短瘫痪持续时间。在发作间期,一是要避免发作诱因,如温度过高或低、拥挤的人群、特殊气味、不规则睡眠、沐浴、洗澡、荡秋千、接触荧光、食用巧克力等;二是长疗程的药物治疗,目前最常用的药物是氟桂利嗪,其作用机制尚不明确,推测可能与其能阻断钙离子通道相关。有报道一半以上患儿在治疗后偏瘫发作的频率、严重程度及发作持续时间有所改善[2]。托吡酯可作为氟桂利嗪治疗无效的后备药物,可单用或联合用药。有报道6例氟桂利嗪治疗无效的病例改用托吡酯治疗6个月后,偏瘫发作频率及发作持续时间均明显减少[15]。本研究的例2患儿,在使用托吡酯治疗癫痫时,偏瘫发作频率亦减半,提示托吡酯对交替性偏瘫有效。需要注意的是,长疗程预防用药虽然可以减少偏瘫发作频率及持续时间,提高患儿生活质量,但是对于远期认知功能是否能起到改善作用,还不能确定[2,5]。AHC患儿远期往往都会存留较严重的认知功能障碍。
综上, AHC常在出生后18个月内起病,以反复偏瘫发作、眼球运动异常及肌张力不全为主要表现的疾病,远期常存在永久性的认知功能损害。临床上ACH容易被漏诊或误诊,ATP1A3基因检测有助于确诊(特别是不典型病例)及遗传咨询。目前,ACH尚无特异性治疗方法,未来基于基因的治疗方法(如基因转入)可能为彻底治愈此病带来希望。
[1] Verret S, Steele JC.Alternating hemiplegia in childhood:a report of eight patients with complicated migraine beginning in infancy [J].Pediatrics, 1971, 47(4):675-680.
[2] Sweney MT, Silver K, Gerard-Blanluet M, et al.Alternating hemiplegia of childhood:early characteristics and evolution of a neurodevelopmental syndrome [J].Pediatrics, 2009, 123(3):e534- e541.
[3] Heinzen EL, Swoboda KJ, Hitomi Y, et al.De novo mutations in ATP1A3 cause alternating hemiplegia of childhood [J].Nat Genet, 2012, 44(9):1030-1034.
[4] Rosewich H, Thiele H, Ohlenbusch A, et al.Heterozygous de-novo mutations in ATP1A3 in patients wim alternating hemiplegia of childhood:a whole-exome sequencing gene identi fi cation study [J].Lancet Neurol, 2012, 11(9):764-773.
[5] Neville BG, Ninan M.The treatment and management of alternating hemiplegia of childhood [J].Dev Med Child Neurol, 2007, 49(10):777-780.
[6] Bourgeois M, Aicardi J, Goutieres F.Alternating hemiplegia of childhood [J].J Pediatr, 1993, 122(5 Pt 1):673-679.
[7] Mikati MA, Kralner U, Zupanc ML, et a1.Alternating hemiplegia of childhood:clinical manifestations and long term outcome [J].Pediatr, 2000, 23(2):134-141.
[8] 高静, 迟兆富, 尚伟, 等.家族性儿童交替性偏瘫一家系报告[J].中华神经科杂志, 2005, 38(12):758.
[9] de Vries B, Stam AH, Beker F, et a1.CACNAlA mutation linking hemiplegic migraine and alternating hemiplegia of childhood [J].Cephalalgia, 2008, 28(8):887-891.
[10] Dobretsov M, Stimers JR.Neuronal function and alpha3 isoform of the Na+/K+-ATPase [J].Front Biosci, 2005, 10:2373-2396.
[11] de Carvalho Agniar P, Sweadner KJ, Penniston JT, et a1.Mutations in the Na+/K+ -ATPase alpha3 gene ATP1A3 are associated with rapid-onset dystonia Parkinsonism [J].Neuron, 2004, 43(2):169-175.
[12] Roubergue A, Ruze E, Vuillaumier-Barrot S, et a1.The multiple faces of the ATP1A3-related dystonic movement disorder [J].Mov Disord, 2013, 28(10):1457-1459.
[13] Ishii A, Saito Y, Mitsui J, et a1.Identification of ATPlA3 mutations by exome sequencing as the cause of alternating hemiplegia of childhood in Japanese patients [J].PLoS One, 2013, 8(2):e56120.
[14] 杨小玲, 张月华, 袁大伟, 等.儿童交替性偏瘫ATPlA3基因突变特点及其对不典型病例的诊断 [J].中华儿科杂志, 2015, 53(11):835-839.
[15] 江文静, 杜滨锋, 迟兆富, 等.托吡酯治疗儿童交替性偏瘫的疗效观察[J].临床神经病学杂志, 2012, 23(3):212-213.
ATP1A3 mutations in childhood alternating hemiplegia:a report of two cases with literature review
ZHANG Ting, MA Jiannan, XIAO Nong (Children's Hospital of Chongqing Medical University, Ministry of Education Key Laboratory of Child Development and Disorders, China International Science and Technology Cooperation Base of Child Development and Critical Disorders, Chongqing Key Laboratory of Pediatrics, Chongqing 400014, China)
ObjectiveTo explore clinical manifestations, genetic diagnosis and treatment of childhood alternating hemiplegia.MethodsTwo patients were clinically diagnosed as alternating hemiplegia.ATP1A3 gene sequencing was performed on these two children and their parents.Literatures on childhood alternating hemiplegia were reviewed.ResultsBoth patients were female.The fi rst symptom of the fi rst girl was alternating hemiplegia occurring at 4-month-old.For the second girl, the fi rst symptom was seizure occurred at 6-month-old, and the typical symptoms including alternating hemiplegia occurred at the second year of course.Heterozygous missense mutations of c.2401G>A (p.D801N) and c.2731G>C (p.A911P) were found in ATP1A3 gene of these two girls, the latter hasn’t been reported in the Human Gene Mutation Database (HGMD) Professional.ConclusionsATP1A3 gene sequencing should be conducted for children clinically diagnosed as alternating hemiplegia, which has important signi fi cance for diagnosis and genetic counseling.
alternating hemiplegia; ATP1A3 gene; child
10.3969/j.issn.1000-3606.2017.02.013
2016-12-08)
(本文编辑:邹 强)
肖农 电子信箱:xiaonongwl@163.com
猜你喜欢
健康体检与管理(2022年4期)2022-05-13
全科护理(2022年3期)2022-02-18
中国康复(2021年6期)2021-11-30
中国生殖健康(2020年2期)2021-01-18
小学生导刊(2018年13期)2018-06-29
中国生殖健康(2018年2期)2018-01-12
中国现代神经疾病杂志(2017年1期)2017-03-29
中国康复理论与实践(2015年10期)2015-12-24
中国神经免疫学和神经病学杂志(2014年5期)2014-05-08
