
微藻基因工程概述*
2017-05-12 07:16闫晋飞杨玉莹马淑霞
生物学通报 2017年4期
闫晋飞 杨玉莹 马淑霞
(1国家海洋局天津海水淡化与综合利用研究所 天津 300192 2沈阳药科大学生命科学与生物制药学院 辽宁沈阳 110016)
微藻是一种单细胞真核生物,是地球上出现最早的物种之一,广泛分布于海洋、淡水湖泊等水域。全球已鉴定的微藻种类达2万余种。微藻丰富的代谢产物中,包含其代谢特有的一些种类的多糖、多不饱和脂肪酸(PUFAs)、生物活性肽、蛋白质、抗氧化剂、免疫活性因子等。这些代谢产物在食品、医药、科研、能源等行业具有重要应用价值。因此,如何通过遗传转化和基因编辑技术修饰微藻基因和引入外源基因,从而提高微藻相关代谢产物的产量,成为国内外学者竞相研究的热点。
随着二代测序成本的降低,一些藻类基因组先后被公布,这为针对微藻的遗传学改造提供了更加清晰的背景,一定程度上推动了微藻基因工程技术的发展。本文将对现今应用于微藻的遗传介导体系及基因编辑技术进行简单总结。
1 真核微藻常用的遗传介导转化方法
细胞核、线粒体和叶绿体是微藻细胞内主要存在的3套遗传体系。基于微藻细胞器膜及其线粒体和叶绿体的存在,可选用基因枪法将外源目的基因导入其线粒体和叶绿体中并实现瞬时表达。将外源基因导入微藻细胞核中的常用方法有:
1.1 玻璃珠法 玻璃珠法(glass beads method),又叫 PEG(聚乙二醇)法,由于聚乙二醇能强烈刺激原生质体吸收 DNA、RNA等大分子物质,因此将玻璃珠、聚乙二醇、外源基因加入微藻细胞中,在一定条件下混合搅拌,可转入外源基因。该法成本低,转化率高,操作简单易行,但具有一定的宿主局限性,目前仅能用于衣藻[1]和杜氏盐藻等缺壁的藻株中,尚未广泛应用。
1.2 碳化硅细丝转化法 碳化硅细丝转化法(silicon carbide whiskers),也叫金刚砂法,最早在1993年应用于莱茵衣藻[2]的转化,该法与玻璃珠法类似,不同之处在于它使用碳化硅细丝代替玻璃珠。由于碳化硅细丝的使用,藻株可以不用去除细胞壁,其宿主范围变广,但是碳化硅细丝价格较高且不易购买,因而也未能得到广泛使用。
1.3 电击法 电击法(electroporation),也称电穿孔法,是利用高压使细胞膜通透性瞬间增大,细胞吸收周围外源基因,从而实现基因导入的一种方法。电击法操作较简单,有较为标准的实验流程,不足之处是耗材成本相对较高。目前此方法被大多数实验室所采用,主要用于酵母和裂壶藻等相对细胞壁较厚、个体相对较小的真核微生物转化中,此外,在绿藻门、硅藻门[3]、褐藻门及红藻门[4]的一些种属均有应用电击法的报道。
1.4 粒子轰击法 粒子轰击法(particle bombardment),也叫基因枪法,是指在真空条件下,用外源DNA包裹金属颗粒(金粒或钨粒),利用基因枪装置产生的高压氦气冲击波加速微弹,使其直接穿透细胞壁和细胞膜,从而使外源DNA进入细胞并整合到细胞染色体中,最终实现稳定遗传和表达的目的。该方法对靶细胞来源基本无限制,应用面较广,操作简单,转化时间短、频率高;其缺点是需要专门的实验设备,实验耗材成本是以上所提到的转化方法中最高的。现主要应用于水稻、拟南芥等高等植物的瞬时转化验证实验中,在微藻中,已成功应用该技术的有莱茵衣藻、小球藻[5]、团藻、三角褐指藻、雨生红球藻[6]等。
1.5 农杆菌转化法 农杆菌转化法(Agrobacteriummediated transformation)通常用于高等植物细胞中。某些农杆菌的细胞含有 Ti(Tumour inducing)质粒或Ri(Root inducing)质粒,其上有一段T-DNA(Transferring DNA),农杆菌侵染植物组织后进入靶细胞,可将T-DNA整合入靶细胞基因组中,并通过减数分裂稳定遗传给后代。随着农杆菌转化法的改进与推广,科学家尝试将其用于微藻转化。Kumar等[7]首先成功地用该方法转化莱茵衣藻,获得了较高的转化效率,证实了该方法应用于微藻转化的可行性。此后,各国学者尝试将其用于一系列其他微藻中,例如淡水的雨生红球藻、普通小球藻[8]、斜生栅藻及海水藻如紫菜、杜氏盐藻[9]、裂壶藻等,取得了一定效果。农杆菌转化法成本低廉,效率较高,但该方法在如何制备不同藻类的原生质体、确定农杆菌与目标藻体的比例、共培养时长等问题上存在挑战,不同藻类的操作流程不尽相同,需进行实验探索,一定程度上限制了其普及。
此外,病毒的转染也是将外源基因导入细胞的一种方法,此法主要用于人及哺乳类动物细胞的基因改造,但用此法对微藻进行外源基因的转化未见报道,这可能是由于目前对微藻的研究尤其是遗传背景研究等方面不全所致。
2 发展中的基因编辑系统
基因编辑是对基因组中靶DNA序列精确修改的一种技术,在后基因组时代,已成为研究微藻基因功能最直接有效的方法之一。目前微藻基因功能的研究主要依赖于基因敲除和外源基因表达技术,从失去功能和获得功能正、反2个方面解析基因功能。近年来,基因编辑技术不断发展,从传统的基因插入突变和 RNA干扰技术,发展到一些新兴的序列特异性核酸酶技术都能一定程度上有效地编辑基因。
2.1 传统基因敲除方法 研究人员应用传统基因敲除方法对部分微藻进行靶向DNA修饰。主要有以下3种技术:随机插入突变、同源重组和RNA干扰。
2.1.1 随机插入突变 理论上,大规模的随机插入突变可在基因组内敲除任一基因。因而,利用逆转录病毒、农杆菌等介导,在靶细胞的基因组中随机插入目的基因,建立一个携带随机插入突变的细胞库,然后通过相应的标记筛选以获得所需要的基因敲除细胞。基于随机插入突变的基因捕获技术有效率高、能使靶基因完全失活且容易分离鉴定的优点。利用该法进行基因敲除所用的载体和方法随受体的不同有所不同:逆转录病毒一般用于动、植物细胞,农杆菌介导的 T-DNA转化和转座子法常用于植物细胞,噬菌体主要用于细菌,基因枪法可用于微藻例如三角褐指藻[10]突变体文库的构建。目前,受微藻自身生物学特性和遗传特性的影响,加之插入标签载体会在很大程度上影响研究的进程与结果,微藻随机插入突变的研究主要局限于少数模式微藻。
2.1.2 同源重组技术 利用同源重组(homologous recombination,HR)原理进行基因敲除,即构建含有目的基因和靶基因同源片段的重组载体,将其导入靶细胞内,通过同源序列间的重组交换,将外源目的基因整合进受体基因组的目标位置上,以敲除靶基因,并表达外源目的基因,通过研究外源目的基因插入前、后生物体或靶细胞遗传特性的改变研究基因功能。
基于HR的基因敲除技术分为完全型基因敲除和条件型基因敲除。完全型基因敲除指利用HR原理完全消除细胞或动物个体中的靶基因活性;条件型基因敲除是指将基因的修饰限制在细胞发育的特定阶段或某种特定细胞的基因敲除,应用较广泛的条件型基因敲除系统有Cre/Lox系统[11]、FLP/FRT系统。目前微藻研究中完全型基因敲除应用最为广泛,条件型基因敲除应用鲜见报道。
2.1.3 RNA干扰技术 RNA干扰(RNA interference,RNAi)是转录后水平的基因沉默机制,是siRNA(small interfering RNA,长 20~25 bp的双股 RNA)分子在mRNA水平上诱发的同源mRNA高效特异性降解的现象,于1998年由Fire首次发现并命名。该方法将短片段的siRNA分子导入细胞内,特异性地降解细胞内与其同源的mRNA,从而阻断特定基因的表达,以达到干扰靶基因表达的目的。RNAi操作简单,成本低,效率高,特异性强,稳定性高,应用较广泛。但由于siRNA不能有效结合所有基因的mRNA,将靶基因全部剔除,只是降低基因的表达水平,产生不完全敲除且具有临时性等,应用受到限制。RNAi技术作为基因敲除的一种重要方法,在动、植物研究中应用广泛,在微藻中的应用常见于莱茵衣藻[12]。
2.2 新兴的基因编辑技术 序列特异性核酸酶(sequence-specific nucleases,SSN)技术通过引入SSN在基因组特定位点造成DNA双链断裂,从而激活细胞天然的“同源重组”及“非同源末端连接(non-homologous end joining,NHEJ)”DNA 修复机制的定向发生,实现基因组定向遗传修饰效率的大幅提升。迄今为止,在基因组定向遗传修饰研究及应用领域,已经有多种不同类型的序列特异性核酸酶被有效使用[13]。下面代表性地介绍几种在微藻中具有应用潜力的SSN技术。
2.2.1 锌指核酸酶基因打靶技术 锌指核酸酶(zinc-finger nuclease,ZFNs)是一种人工合成的限制性内切酶,其核心设计理念是将2个具有特定功能的结构域,特异性识别DNA序列的锌指结构域和非特异核酸内切酶活性的DNA切割域,即特异性识别模块与功能模块之间相互融合,形成具有特定基因编辑功能的蛋白。
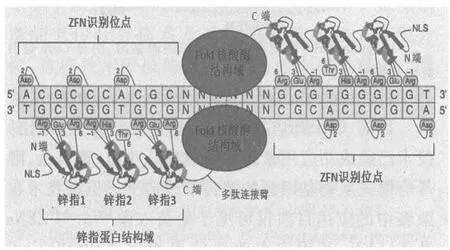
图1 ZFNs技术示意图[14]
锌指核酸酶N末端是锌指蛋白DNA的结合域,由一系列 Cys2-His2锌指蛋白(zinc-fingers,ZF)串联组成(一般 3~4个),每个锌指可特异识别并结合DNA链上的3个连续碱基;C末端多为非特异性FokⅠ核酸酶结构域,FokⅠ是一种type IIS型的核酸内切酶,源于限制性内切酶FokⅠ的一个亚基,二聚化后才具有切割活性。实际应用时,通常针对靶序列设计一对ZFNs,并在靶序列之间留有5~7 bp的间隔区域(spacer)以确保一对ZFN中FokⅠ二聚体的形成[13]。FokⅠ二聚体化后产生酶切功能,并在此特异位点产生1个DNA双链切口(double strands breaks,DSB),细胞通过 NHEJ机制修复DSB。在修复重连过程中,碱基随机的插入或丢失造成移码突变,使基因无法正常表达,实现基因敲除;当存在含有同源序列的重组供体时,发生HR修复,则实现外源基因的定点敲入。
2013 年,Sizova 等[15]首次将 ZFNs技术应用于莱茵衣藻,从而实现了高效率的基因定点修饰。以ZFNs为介导的基因敲除技术可精确修饰靶基因或其周围的调控元件,但也存在应用上的难题:1)如何设计、筛选具有高亲和性及高特异性的锌指蛋白酶;2)ZFN可能会对基因组非靶向位点随机切割,损伤细胞;3)如何有效调控ZFNs的表达等。
2.2.2 TALENs技术 转录激活因子样效应物核酸酶(transcription activator-like effector nuclease,TALENs)靶向基因敲除技术,其设计和构建的原理是植物病原体黄单胞菌(Xanthomonas)分泌的一种转录激活子样效应因子(transcription activator-like effector,TALE)可以识别特定的DNA序列。
与 ZFNs相类似,TALENs主要包括 2个组成部分:DNA识别域(TALE蛋白)和剪切结构域(FokⅠ核酸内切酶)。TALE蛋白(如图 2A)由17~18个特异性识别DNA的串联重复基本单元构成,每个串联重复基本单元由34个氨基酸残基构成,其中第12和13位残基是靶向识别的关键位点,称作重复可变氨基酸残基位点(repeat variant diresidues,RVDs),不同 RVDs组合能特异识别一个碱 基 (HD →C、NI→A、NG →T、NN →G、MN →G或A)。利用TALE这种识别单元与核苷酸碱基恒定的对应关系,实际操作中可通过靶位点的DNA序列反推二联氨基酸序列构建TALE靶点识别模块。目前TALENs的构建中最常用的是Cermak等发表的“Golden Gate”,与其类似的构建方法都遵循该思路:将4种碱基对应的TALE模块的DNA序列连接到载体上,然后通过酶切连接将各TALE单体组成阵列,最后与N端的核定位信号(nuclear localization signal,NLS)及 C 端的 FokⅠ核酸酶连接起来,完成 TALENs 表达质粒的构建[16]。 TALENs质粒共转入细胞后,其表达的融合蛋白在NLS帮助下由细胞质进入细胞核,分别找到其靶基因位点并与靶位点特异结合。此时,2个TALENs融合蛋白中的FokⅠ功能域形成二聚体并发挥非特异性内切酶活性,于2个靶位点之间剪切目的基因,产生1个DSB。由此,可以在该位点进行敲入(knock-in)、敲出(knock-out)或点突变等操作。
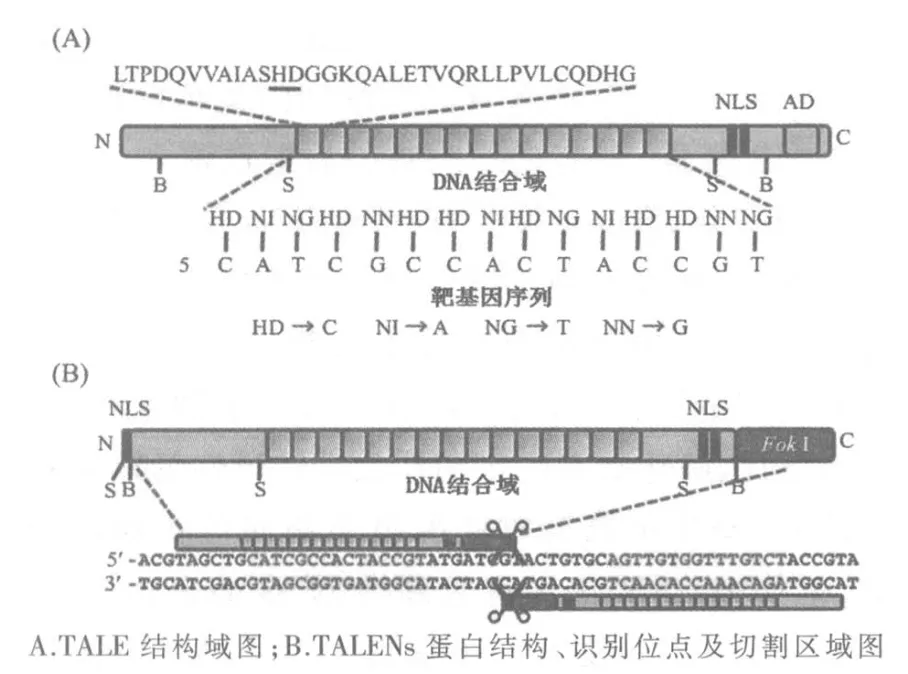
图2 TALENs技术示意图[17]
TALENs作为新兴的基因定点编辑工具,近几年已成功应用于动、植物和酵母[17]等微生物细胞,在微藻的莱茵衣藻[18]及三角褐指藻[19]中也已得到运用。它成功解决了ZFNs技术识别目标基因序列方面的低特异性及易受上、下游序列影响等问题,同时具有与其相等甚至更好的活性,无基因序列、细胞、物种限制,载体构建简单易行,成本低,脱靶率小,细胞毒性小。目前 TALENs技术的不足之处主要在于2个方面:1)设计识别仅20个碱基序列的 TALE核酸酶可能含有1 000多个氨基酸,分子量过大易引起细胞免疫反应,减弱TALE在细胞中的作用;2)与ZFNs相比,脱靶问题虽然得到了一定控制但依然存在。
2.2.3 CRISPR/Cas系统 2013年以来,研究人员在包括Science和Nature Biotechnology等著名杂志上发表多篇文章介绍CRISPR(clustered regularly interspaced short palindromic repeats) /Cas(CRISPR-associated)系统。天然的CRISPR/Cas系统广泛存在于细菌及古生菌中,是机体长期进化形成的RNA指导的降解入侵病毒、噬菌体DNA的适应性免疫系统。Cas基因家族编码的蛋白具有与核酸结合及核酸酶、聚合酶、解旋酶等活性,根据其基因核心元件序列的不同[20],CRISPR/Cas免疫系统可分为Ⅰ型、Ⅱ型、Ⅲ型3种类型。其中,Ⅰ型和Ⅲ型的CRISPR/Cas免疫系统需要多种Cas蛋白形成复合体才能切割DNA双链,而Ⅱ型CRISPR/Cas免疫系统只需要1个Cas9蛋白即可切割DNA双链。
CRISPR/Cas9是目前研究最为充分的系统,其主要工作结构包括Cas9蛋白、crRNA、tracrRNA 3个部分。Cas9是由1 409个氨基酸组成的多结构域蛋白,含有2个核酸酶结构域:位于蛋白中间位置的HNH核酸酶结构域及氨基端的RuvC-like结构域。当外源DNA入侵细胞,crRNA经RNaseⅢ催化和tracrRNA形成双链RNA结构介导Cas9蛋白定向切割与crRNA互补的靶序列位点[16]。研究发现,将crRNA与tracrRNA结合设计形成一条单链RNA(single guide RNA,sgRNA)同样也能指导Cas9蛋白切割双链DNA,这样进一步提高了操作的简便性。如图3所示,在sgRNA的指导下,Cas9蛋白HNH核酸酶结构域剪切与sgRNA互补配对的模板链,RuvC-like结构域可以对另一条链进行切割,从而形成DSB。其中切割位点位于原型间隔序列毗邻基序(protospacer adjacent motif,PAM)上游 3~8 nt处。

图3 CRISPR/Cas9识别目标基因组DNA示意图[21]
目前,CRISPR/Cas9靶向基因改造(敲入、敲出)技术已经成功应用于拟南芥、烟草、小鼠、斑马鱼、酵母等模式生物基因组的遗传学改造上。但该技术在微藻中的应用目前仅可见于莱茵衣藻[22]。与ZFNs及TALENs基因定点改造技术相比,CRISPR/Cas9系统具有显著优点:无基因序列、细胞、物种限制;可以对所有后面紧随 NGG(PAM)的 20 bp的基因序列进行编辑,理论上在基因组上8 bp就有一个适合CRISPR/Cas9编辑的位点,而对于ZFNs和TALENs分别需要500 bp和125 bp才会有合适的编辑位点[23—24];可同时作用于多个靶位点;载体构建简单且Cas9基因相同,针对特定物种特定基因位点只需找到该物种对应的高效启动子及设计识别靶基因的sgRNA,实验周期短、成本低,而ZFNs或TALENs对任一基因位点编辑时都需要设计和组装2种核酸酶;另外,该技术还可以通过向受体直接导入sgRNA/Cas9 mRNA或sgRNA/Cas9蛋白的形式,实现目的基因的敲除及编辑,目前相关产品已商品化,使用方便。当然,该方法的普及仍存在一些技术难题,例如脱靶效应、怎样将Cas9和gRNAs呈递给成体组织细胞、受体启动子的选择等。总体而言,CRISPR/Cas系统正快速超越ZFNs和TALENs等其他编辑工具,成为微藻研究及其他生物研究的重要技术。
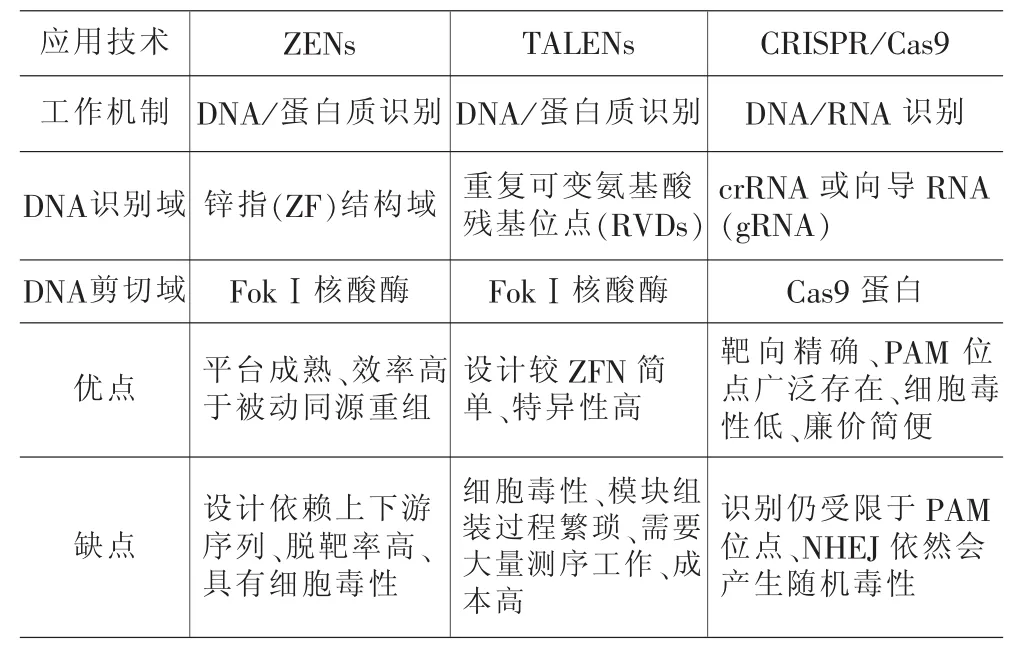
表1 3 种基因编辑技术的对比[14,16,23]
3 小结
微藻遗传背景的逐渐清晰,以及现代生物学研究技术的不断发展,使得对各种微藻进行基因工程学改造成为可能。本文对目前应用于微藻改造的遗传介导体系及基因编辑技术做了简要介绍,这些技术手段的运用,将为原本在微藻中产量基微,提取困难、不易被直接利用的一些生物活性物质的开发与应用提供广阔的空间。
主要参考文献
[1]黄非,黄秦,黄敏,等.莱茵衣藻玻璃珠法快速转化检测体系的建立.四川大学学报(自然科学版),2008,45(3):694.
[2]Dunahay T G.Transformation ofChlamydomonas reinhardtiiwith silicon carbide whiskers.Biotechniques,1993,15(3):452.
[3]Chen H L,Li S S,Huang R,et al.Conditional production of a functional fish growth hormone in the transgenic line ofNannochloropsis oculata(Eustigmatophyceae)1.Journal of Phycology,2008,44(3):768.
[4]Kübler J E,Minocha S C,Mathieson A C.Transient expression of the GUS reporter gene in protoplasts ofPorphyra miniata(Rhodophyta).Marine Biotechnology,1994(1):165.
[5]Dawson H N,Burlingame R,Cannons A C.Stable transformation ofChlorella:rescue of nitrate reductase-deficient mutants with the nitrate reductase gene.Current Microbiology,1997,35(6):356.
[6]Teng C,Qin S,Liu J,et al.Transient expression of lacZ in bombarded unicellulargreen algaHaematococcuspluvialis.Journal of Applied Phycology,2002,14(6):497.
[7]Kumar S V,Misquitta R W,Reddy V S,et al.Genetic transformation ofthegreen alga—ChlamydomonasreinhardtiibyAgrobacterium tumefaciens.Plant Science,2004,166(3):731.
[8]San Cha T,Yee W,Aziz A.Assessmentof factorsaffecting Agrobacterium-mediated genetic transformation of the unicellular green alga,Chlorella vulgaris.World Journal of Microbiology and Biotechnology,2012,28(4):1771.
[9]Anila N,Chandrashekar A,Ravishankar G A,et al.Establishment ofAgrobacterium tumefaciens-mediated genetic transformation inDunaliella bardawil.European Journal of Phycology,2011,46(1):36.
[10]刘飞飞,李秀波,方仙桃,等.三角褐指藻产油突变株的筛选.水生生物学报,2013,37(4):799.
[11]Ma B G,Duan X Y,Niu J X,et al.Expression of stilbene synthase gene in transgenic tomato using salicylic acid-inducible Cre/loxP recombination system with self-excision of selectable marker.Biotechnology Letters,2009,31(1):163.
[12]Hu J,Deng X,Shao N,et al.Rapid construction and screening of artificial microRNA systems inChlamydomonas reinhardtii.The Plant Journal,2014,79(6):1052.
[13]韦韬,张勇,刘玉,等.序列特异性核酸酶及其在植物基因组定向修饰中的应用.中国细胞生物学学报,2013(11):14.
[14]Porteus M H,Carroll D.Gene targeting using zinc finger nucleases.Nature Biotechnology,2005,23(8):967.
[15]Sizova I,Greiner A,Awasthi M,et al.Nuclear gene targeting inChlamydomonasusing engineered zinc-finger nucleases.The Plant Journal,2013,73(5):873.
[16]Ma D,Liu F.Genome Editing and Its Applications in Model Organisms.Genomics,Proteomics&Bioinformatics,2015(13):336.
[17]Aouida M,Li L,Mahjoub A,et al.Transcription activatorlike effector nucleases mediated metabolic engineering for enhanced fattyacidsproduction inSaccharomyces cerevisiae.Journal of Bioscience and Bioengineering,2015,120(4):364.
[18]Gao H,Wright D A,Li T,et al.TALE activation of endogenous genes inChlamydomonas reinhardtii.Algal Research,2014(5):52.
[19]Weyman P D,Beeri K,Lefebvre S C,et al.Inactivation ofPhaeodactylum tricornutumurease gene using transcription activator-like effector nuclease-based targeted mutagenesis.Plant Biotechnology Journal,2015,13(4):460.
[20]李君,张毅,陈坤玲,等.CRISPR/Cas系统:RNA靶向的基因组定向.Hereditas(Beijing),2013,35(11):1265.
[21]Mali P,Yang L,Esvelt K M,et al.RNA-Guided Human Genome Engineering via Cas9.Science,2013,339(6121):823.
[22]Baek K,Kim D H,Jeong J,et al.DNA-free two-gene knockout inChlamydomonas reinhardtiiviaCRISPR-Cas9 ribonucleoproteins.Scientific Reports,2016.doi:10.1038/srep306206.
[23]Wang H,Yang H,Shivalila C S,et al.One-step generation of mice carrying mutations in multiple genes by CRISPR/Casmediated genome engineering.Cell,2013,153(4):910.
[24]李聪,曹文广.CRISPR/Cas9介导的基因编辑技术研究进展.生物工程学报,2015(11):1531.
猜你喜欢
舰船科学技术(2022年11期)2022-07-15
古今农业(2022年1期)2022-05-05
无机化学学报(2020年7期)2020-07-20
中国预防兽医学报(2020年2期)2020-06-01
三农资讯半月报(2020年8期)2020-05-13
西藏农业科技(2019年3期)2019-11-04
现代园艺(2018年3期)2018-02-10
环境保护与循环经济(2017年3期)2017-09-26
上海农业学报(2017年3期)2017-04-10
中国病理生理杂志(2017年2期)2017-01-17
