
结肠癌淋巴结转移基因谱表达差异分析
2017-06-19 19:22赵志丹刘建华钟白云王恝歆谢婷彦张秋焕冯斯斯邓辉
临床检验杂志 2017年5期
赵志丹,刘建华,钟白云,王恝歆,谢婷彦,张秋焕,冯斯斯,邓辉
(中南大学湘雅医院检验科,长沙 410008)
·研究生园地·
结肠癌淋巴结转移基因谱表达差异分析
赵志丹,刘建华,钟白云,王恝歆,谢婷彦,张秋焕,冯斯斯,邓辉
(中南大学湘雅医院检验科,长沙 410008)
目的 探讨人结肠癌细胞系SW480、SW620基因谱的表达差异。方法 针对NCBI的GEO数据库中编号为GDS756的基因芯片表达数据,用GSEA软件进行基因集富集分析及领头亚群分析,利用FunRich分析软件针对SW480、SW620细胞领头亚群基因进行验证性功能注释,STRING在线分析系统对显著性富集基因集领头亚群基因进行生物网络分析,获得SW480、SW620细胞中心节点基因,并将中心节点基因与高重叠领头亚群基因进行联合分析,以获得参与多功能的中心节点基因。结果 GSEA软件分析筛选出SW480细胞显著性富集基因集12个,领头亚群基因491个,高重叠基因7个;SW620细胞显著性富集基因集80个,领头亚群基因870个,高重叠基因6个。对显著性富集基因集领头亚群基因进行生物网络分析筛选出SW480、SW620细胞相关中心基因数目分别为5个及8个;结合GSEA及生物网络分析结果,获得SW620细胞2个参与多功能调控的核心基因TOP2A、CDK1。结论 遗传背景相同的结肠癌SW620细胞与SW480细胞具有不同的基因功能分布特点,SW620细胞多功能中心节点基因TOP2A、CDK1与结肠癌发展相关信号通路高度相关。
SW480;SW620;基因表达谱;基因集富集分析;生物网络
结肠癌的淋巴结转移是患者5年生存率降低的重要原因[1]。目前认为,结肠癌淋巴结转移是一个多基因共同作用的过程,但其具体机制仍未明确。本研究利用结肠癌原发灶SW480细胞与结肠癌淋巴结转移SW620细胞基因芯片表达谱数据,联合应用基因集富集分析(GSEA)与差异基因生物网络分析。探讨结肠癌细胞从侵袭到转移这一过程中的基因表达谱的差异。以期加深对结肠癌侵袭、转移机制差异的认识,为进一步的分子生物学研究提供新的实验依据。
1 材料与方法
1.1 获取基因芯片表达谱数据 登陆美国国立生物信息中心(NCBI)基因表达共享数据库GEO(Gene Expression Omnibus, https://www.ncbi.nlm.nih.gov/geo/)。获取系列号为GDS756的基因表达谱数据集。该数据集包含原位结肠癌细胞(SW480)、淋巴结转移灶结肠癌细胞(SW620)的基因表达谱数据集。SW480的3次独立重复芯片分析结果编号为GSM21712、GSM21713、GSM21714;SW620的3次独立重复芯片分析结果编号为 GSM21715、GSM21716、GSM21718。两细胞系来源于同一患者,购自欧洲细胞中心(ECACC, Salisbury, UK)。
1.2 数据预处理 应用Affymetrix Expression Console软件对原始芯片数据进行质控,用RMA算法进行数据的标准化处理。在GEO下载GPL96(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL96)基因平台TEX文本,找到对应探针的基因名称(Gene Symbol),对输出基因进行注释。
1.3 基因集富集分析 本次分析选取了以下7个参照基因集:H:hallmark gene sets(效应特征基因集合),共50组;C1:位置基因集合(positional gene sets),根据染色体位置,共326个;C2:专家共识基因集合(curated gene sets),基于通路、文献等;C3:模式基因集合(motif gene sets),主要包括microRNA和转录因子靶基因两部分;C4:计算基因集合(computational gene sets),通过挖掘癌症相关芯片数据定义的基因集合;C5:基因本体论集合(Gene Ontology gene sets),包括生物学过程(biological process),细胞原件(cellular component)和分子功能(molecular function)3个部分;C6:癌症特征基因集合(oncogenic signatures),大部分来源于GEO未发表芯片数据;涉及广泛生理调控机制。选择default weighted enrichment statistic方法,并设置错误发现率(false discovery rate,FDR)Q<0.25,每次随机选择100次重复分析(Number of permutation=100)、总体错误率(family-wise error rate,FWR)<0.05的基因集作为阳性基因集。
1.4 基因集领头亚群分析 按标准化富集评分(Normalized Enrichment Score,NES)>2.0、FDRQ<0.25、FWR<0.05筛选显著富集基因集,进行领头亚群分析(1eading edge subset analysis)。领头亚群基因是富集评分(Enrichment Score,ES)绝对值从0增加至最大的这段区间内的基因集合。由于SW480、SW620领头亚群基因的最高重叠次数不同,高重叠基因筛选需进行差异性处理,以保证所得基因在各自细胞中的重要性一致。以0.7×最高重叠次数作为阈值,重叠次数大于该阈值为标准,分别筛选SW480细胞领头亚群中的高重叠基因及SW620细胞领头亚群中的高重叠基因。
1.5 领头亚群基因功能注释 利用FunRich分析软件[2]针对SW480、SW620细胞针对领头亚群基因进行生物学过程(Biological process)功能富集、信号通路(Biological pathway)注释。针对每个功能富集分析和信号转导通路富集分析得到领头亚群基因在功能集中的总数,以及FunRich数据库中基因在功能集中的总数,利用超几何检验计算显著性程度(P值),再通过BH纠正P值来降低假阳性,以P<0.01为显著性阈值,分别得到具有统计意义的高频率注释、信号通路;选取存在差异的前12个生物学过程进行对比分析。
1.6 领头亚群基因的生物网络分析 STRING 10.0生物网络的生成主要基于:(1)已有实验性数据,(2)文献报道,(3)生物信息学分析预测。将1.4中所得领头亚群基因合并提交STRING 10.0网络软件(http://string-db.org/),构建蛋白质相互作用网络。计算网络中各节点的度值(d),分别对SW480、SW620结果进行差异性处理,以0.7×最高度值作为阈值,d大于该阈值为标准,分别筛选SW480细胞领头亚群中的中心节点以及SW620细胞领头亚群中的中心节点。
1.7 领头亚群高重叠基因与中心节点基因的联合分析 将SW480细胞领头亚群分析所得高重叠基因与领头亚群基因生物网络分析所得中心节点基因取交集,筛选出高重叠基因中所含的中心节点基因;同理可筛得SW620细胞高重叠基因中所含的中心节点基因。
2 结果
2.1 GSEA富集的基因集 有4 917个基因集在SW480细胞中表达上调,其中205个FDRQ<0.25,722个P<0.05,339个P<0.01。5 014个基因集在SW620细胞中表达上调,其中1 310个FDRQ<0.25,1 337个P<0.05,861个P<0.01。
按NES>2.0、FDRQ<0.25、FWR<0.05筛选显著富集基因集,筛选出12个SW480细胞显著富集基因集和80个SW620细胞显著富集基因集。在SW480细胞显著富集基因集中,细胞增殖相关基因集7个,上皮间质转化(EMT)相关基因集3个,肿瘤微环境相关基因集1个,角蛋白相关的基因集1个。SW620细胞前20个显著富集基因集中,关于细胞周期的基因集有7个,关于细胞增殖的基因集有6个,关于肿瘤转移的基因集有4个,与肿瘤患者预后相关的基因集有2个,与RNA剪切相关基因集有1个。见表1。
2.2 GSEA领头亚群分析结果 SW480细胞12个显著富集基因集中,分析得到491个领头亚群基因,筛选出7个至少参与3个基因集领头亚群基因(KRT5、SLC2A3、KRT81、KRT13、SPINT2、S100A2、LCN2);SW620细胞80个显著富集基因集中,分析得到870个领头亚群基因,筛选出6个至少参与35个基因集领头亚群基因(CDK1、RRM1、ZWINT、TOP2A、MCM2、KIF11)。两细胞领头亚群基因无交集。

表1 SW480、SW620显著富集基因集生物学功能
2.3 领头亚群基因的功能标注 按生物学过程(Biological process)标签标注两领头亚群基因,选取存在差异的前12个生物学过程进行对比分析。见图1。SW480细胞领头亚群基因中有483个基因具有生物学过程功能注释,其中与细胞生长/存活(cell growth and/or maintenance)关联基因占11.2%(P<0.01)、细胞通讯(cell communication)关联基因占25.1%(P<0.01)、蛋白质代谢(protein metabolism)关联基因占10.1%(P<0.01)、信号转导(signal transduction)关联基因占25.9%(P<0.01)、核酸代谢调控(regulation of nucleobase, nucleoside, nucleotide and nucleic acid metabolism)关联基因占16.4%(P<0.01);SW620细胞领头亚群基因中有858个基因具有生物学过程功能注释,其中与细胞生长/存活(cell growth and/or maintenance)关联基因占6.3%(P<0.01)、细胞通讯(cell communication)关联基因占18.3%(P<0.01)、蛋白质代谢(protein metabolism)关联基因占7.1%(P<0.01)、信号转导(signal transduction)关联基因占19.9%(P<0.01)、核酸代谢调控(regulation of nucleobase, nucleoside, nucleotide and nucleic acid metabolism)关联基因占30.2%(P<0.01)。
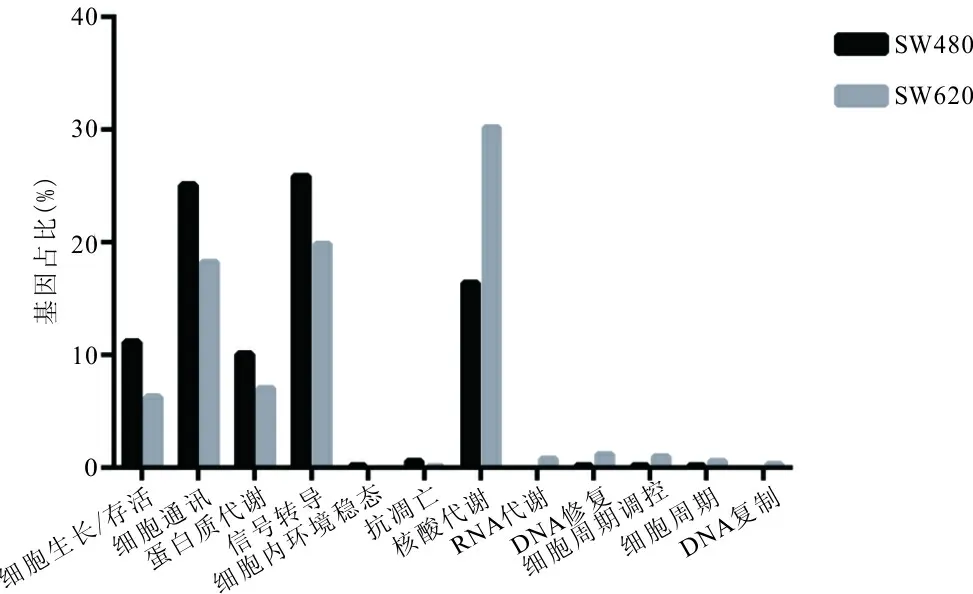
图1 SW480、SW620细胞12个生物学过程比较
按信号通路(biological pathway)标签标注两领头亚群基因,选取存在差异的前12个信号通路进行对比分析,结果发现SW480细胞领头亚群基因中有206个基因具有信号通路功能注释,其中与整合素家族细胞表面相互作用信号通路(integrin family cell surface interactions)关联基因占39.3%(P<0.01)、与Beta1整合素细胞表面相互作用信号通路(beta1 integrin cell surface interactions)关联基因占38.3%(P<0.01)、与IFN-γ信号通路(IFN-gamma pathway)关联基因占36.9%(P<0.01)、蛋白聚糖粘附分子介导信号通路(proteoglycan syndecan-mediated signaling events)关联基因占37.9%(P<0.01)、鞘氨醇磷酸酯调控信号通路(sphingosine 1-phosphate (S1P) pathway)关联基因占36.9%(P<0.01)、PDGF受体信号网(PDGF receptor signaling network)关联基因占36.4%(P<0.01);SW620细胞领头亚群基因中有448个基因具有信号通路功能注释,其中与前体RNA修饰信号通路(processing of capped intron-containing pre-mRNA)关联基因占14.7%(P<0.01)、与mRNA合成信号通路(mRNA processing)关联基因占15.2%(P<0.01)、与mRNA剪切信号通路(mRNA splicing)关联基因占12.5%(P<0.01)、与mRNA剪切核心信号通路(mRNA splicing - major pathway)关联基因占12.5%(P<0.01)、与mRNA转录子成熟信号通路(formation and maturation of mRNA transcript)关联基因占15.4%(P<0.01)、与细胞周期信号通路(cell cycle, Mitotic)关联基因占17.6%(P<0.01)。
2.4 领头亚群基因的生物网络分析 SW480细胞领头亚群相互作用网络由490个结点和1 009条边连接而成。上调基因中d值大于40的中心节点5个,分别为:ITGA2、FOS、CDH1、IL-6、RANBP2。其中ITGA2参与的信号通路包括:整合素家族细胞表面相互作用(integrin family cell surface interactions)、 整合素细胞表面相互作用(beta1 integrin cell surface interactions)、蛋白多糖syndecan介导的信号通路(proteoglycan syndecan-mediated signaling events)、血管内皮生长因子和血管内皮生长因子受体相关信号通路网(VEGF and VEGFR signaling network);FOS、CDH1、IL-6参与所有筛选所得信号通路。
SW620细胞领头亚群相互作用网络由867个结点和8 734条边连接而成。上调基因中d值大于98的中心节点8个,分别为:TOP2A、CDK1、PCNA、PLK1、CCNB1、AURKB、SNRPB、HDAC1。其中TOP2A、SNRPB、HDAC1参与核酸代谢调控(regulation of nucleobase, nucleoside, nucleotide and nucleic acid metabolism);TOP2A、CDK1、PCNA、PLK1、CCNB1、AURKB、HDAC1参与细胞周期(cell cycle)、有丝分裂(mitotic)、DNA复制(DNA replication)相关信号通路;SNRPB参与mRNA加帽(processing of capped intron-containing Pre-mRNA)、mRNA剪切(mRNA splicing)mRNA转录、成熟(formation and maturation of mRNA transcript)、mRNA翻译(gene expression)相关信号通路。
2.5 领头亚群高重叠基因与中心节点基因的联合分析 SW480细胞的领头亚群高重叠基因不含中心节点基因;SW620细胞领头亚群高重叠基因中存在2个中心节点基因:TOP2A、CDK1。根据领头亚群基因功能标注可知TOP2A参与了细胞有丝分裂与核苷酸代谢的调控(regulation of nucleobase, nucleoside, nucleotide and nucleic acid metabolism, cell cycle, mitotic),CDK1参与了细胞周期,DNA复制的调控(cell cycle, mitotic, DNA replication, mitotic M-M/G1 phases)。
3 讨论
生物信息学的快速发展为分子生物研究的大数据分析处理提供了有力支持。基因芯片表达谱分析技术领域中,传统上以差异基因分析为基础的分析方法可以有效筛选样本间明显上调或下调的基因,但容易遗漏部分差异表达不明显但却有重要生物学意义的基因[3]。GSEA的原理是使用预定义的基因集,通常来自功能注释或者先前实验的结果,将基因按两类样本中的差异表达程度进行排序,然后分析预先设定的基因集合是否在这个序列表的顶端或底端富集[4]。GSEA软件分析基因集合而非单个基因的表达变化,因此不容易忽略细微的表达变化,遗漏部分差异表达不显著却有重要生物学意义的基因[5-6]。
Affymetrix Human Genome U133A Array覆盖了33 000多种已经被证实的人类基因,可以对人类基因组中的绝大多数基因表达进行分析。本研究所选取的结肠癌淋巴结转移相关基因芯片GDS756基于以上平台,通过基因集合富集分析得到12个SW480显著富集基因集,80个SW620细胞显著富集基因集;两细胞系基因表达谱存在较大差异(表1)。将领头亚群高重叠基因与中心节点基因取交集筛选多功能节点基因,结果显示SW480的中心节点基因未参与3个以上功能亚群;TOP2A、CDK1作为SW620细胞的中心节点基因,同时参与了35个以上的功能亚群。
拓扑异构酶Topo Ⅱ α 是基因TOP2A的蛋白质产物,在细胞核内催化DNA的断裂重组,可形成断裂、非断裂两种类别的复合体,参与细胞有丝分裂周期的调控,是维持遗传物质稳定性的重要蛋白酶,可作为蒽环类药物的作用靶点[7]。Kang等[8]研究发现TOP2A是PTEN维持细胞核内基因组稳定性的关键靶标,是调控细胞周期的关键靶点。CDK1基因编码细胞周期蛋白依赖性激酶1,可与细胞周期蛋白(Cyclin B1)相结合,二者参与细胞有丝分裂G2/M期调控促进细胞增殖[9-10]。Yang等[11]研究表明,CDK1高表达卵巢癌患者5年生存率较低,抑制CDK1的表达可减弱卵巢癌的增殖能力。TOP2A、CDK1与细胞增殖功能均有密切关系,但在结肠癌淋巴结转移领域的研究相对薄弱仍具有较大的空间。
综上所述,本研究采用生物信息学方法分析已有的结肠癌淋巴结转移芯片数据,低成本、高效地筛选出潜在的结肠癌淋巴结转移相关多功能中心节点基因TOP2A、CDK1。因本研究仅涉及生物信息学分析,TOP2A、CDK1与结肠癌淋巴结转移的相关性仍需在临床样本中进行验证,同时两者参与结肠癌淋巴结转移的具体机制也有待进一步探索。这也是本研究的不足之处及后续的研究方向。
[1]Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016[J]. CA Cancer J Clin, 2016,66(1):7-30.
[2]Pathan M, Keerthikumar S, Ang CS,etal. FunRich: An open access standalone functional enrichment and interaction network analysis tool[J]. Proteomics, 2015,15(15):2597-2601.
[3]Wang CH, Gao XJ, Liao SY,etal. Transcriptome analysis of human breast cancer cell lines MCF-7 and MDA-MB- 435 by RNA-seq[J]. Mol Biol (Mosk), 2015,49(2):279-288.
[4]Debrabant B. The null hypothesis of GSEA, and a novel statistical model for competitive gene set analysis[J]. Bioinformatics, 2017,33(9):1271-1277
[5]Subramanian A, Tamayo P, Mootha VK,etal. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles[J]. Proc Natl Acad Sci USA, 2005,102(43):15545-15550.
[6]冯春琼,邹亚光,周其赵,等.GSEA在全基因组表达谱芯片数据分析中的应用[J].现代生物医学进展, 2009,9(13):2553-2557.
[7]Bau JT, Kang Z, Austin CA,etal. Salicylate, a catalytic inhibitor of topoisomerase II, inhibits DNA cleavage and is selective for the alpha isoform[J]. Mol Pharmacol, 2014,85(2):198-207.
[8]Kang X, Song C, Du X,etal. PTEN stabilizesTOP2Aand regulates the DNA decatenation[J]. Sci Rep, 2015,5:17873.
[9]Eo SH, Kim JH, Kim SJ. Induction of G(2)/M arrest by berberine via activation of PI3K/Akt and p38 in human chondrosarcoma cell line[J]. Oncol Res, 2014,22(3):147-157.
[10]Danielsen SA, Eide PW, Nesbakken A,etal. Portrait of the PI3K/AKT pathway in colorectal cancer[J]. Biochim Biophys Acta, 2015,1855(1):104-121.
[11]Yang W, Cho H, Shin HY,etal. Accumulation of cytoplasmicCDK1 is associated with cancer growth and survival rate in epithelial ovarian cancer[J]. Oncotarget, 2016,7(31):49481-49497.
(本文编辑:许晓蒙)
Differential analysis of gene expression profiles for lymphonode metastasis of colon cancer
ZHAOZhi-dan,LIUJian-hua,ZHONGBai-yun,WANGJia-xin,XIETing-yan,ZHANGQiu-huan,FENGSi-si,DENGHui
(DepartmentofClinicalLaboratoryXiangyaHospital,CentralSouthUniversity,Changsha410008,Hunan,China)
Objective To investigate the differences in the gene expression profiles between SW480 and SW620 cell lines. Methods A dataset of GDS756 containing the gene expression profiles of SW480 and SW620 was downloaded from the GEO database in NCBI. The differential expression genes between SW480 and SW620 were analyzed with gene set enrichment analysis (GSEA) and leading edge subset analysis. The genes in leading edge subset were re-annotated by FunRich software. The core genes of leading edge subset closely relating to SW480 or SW620 were analyzed with the STRING on-line analytical system. The functional core genes closely relating to SW480 or SW620 were obtained by the combined analysis of the core genes and high frequency genes from leading edge subset. Results GSEA identified 12 significantly enriched gene sets, 491 leading edge genes and 7 highly overlapping genes from SW480 and 80 significantly enriched gene sets, 870 leading edge genes and 6 highly overlapping genes from SW620. The STRING system identified 5 core genes from SW480 and 8 from SW620. The combined analysis of GSEA and bionetwork obtained 2 functional core genes,TOP2AandCDK1, from SW620. Conclusion The SW480 and SW620 cells with identical genetic background have different functional gene expression profiles, and the functional core genesTOP2AandCDK1 in SW620 cells may be related to the signal pathways of colon cancer metastasis.
SW480; SW620; gene expression profile; gene set enrichment analysis; bionetwork
10.13602/j.cnki.jcls.2017.05.17
赵志丹,1991年生,男,技师,硕士研究生,主要从事肿瘤学与分子生物学研究。
钟白云,教授,E-mail:zbycsu@hotmail.com。
R735.3+5
A
2017-02-21)
猜你喜欢
中国医药科学(2022年5期)2022-05-05
当代陕西(2018年12期)2018-08-04
西南国防医药(2016年7期)2016-12-01
腹腔镜外科杂志(2016年10期)2016-06-01
中国卫生标准管理(2015年3期)2016-01-14
中国卫生标准管理(2015年1期)2016-01-14
中国当代医药(2015年26期)2015-03-01
现代企业文化(2015年1期)2015-02-28
中国火炬(2014年4期)2014-07-24
中医研究(2014年2期)2014-03-11
