
非综合征型X连锁隐性遗传耳聋家系临床表型及遗传学特征分析
2017-06-24 13:54牛志杰冯永3梅凌云孙捷4陈红胜贺楚峰刘亚兰王雪萍文杰蒋璐
中华耳科学杂志 2017年2期
牛志杰冯永,3梅凌云孙捷,4陈红胜贺楚峰刘亚兰王雪萍文杰蒋璐
1中南大学湘雅医院耳鼻咽喉头颈外科(长沙410008)
2耳鼻咽喉重大疾病研究湖南省重点实验室(长沙410008)
3中南大学医学遗传学国家重点实验室(长沙410078)
4新疆医科大学第一附属医院耳鼻咽喉科(乌鲁木齐830011)
非综合征型X连锁隐性遗传耳聋家系临床表型及遗传学特征分析
牛志杰1,2冯永1,2,3梅凌云1,2孙捷1,2,4陈红胜1,2贺楚峰1,2刘亚兰1,2王雪萍1,2文杰1,2蒋璐1,2
1中南大学湘雅医院耳鼻咽喉头颈外科(长沙410008)
2耳鼻咽喉重大疾病研究湖南省重点实验室(长沙410008)
3中南大学医学遗传学国家重点实验室(长沙410078)
4新疆医科大学第一附属医院耳鼻咽喉科(乌鲁木齐830011)
目的分析一个X连锁隐性遗传性耳聋家系的临床特征及遗传学规律。方法通过问卷调查,收集家系成员临床资料,并进行听力学检测、专科检查及全面体查,对临床听力学特征进行分析并绘制遗传图谱,并对先证者进行GJB2、GJB3以及线粒体全序列进行筛查。结果家系成员共28人,其中男性患者5人,分布于第二、三及四代,耳聋发生于0~5岁,迅速进展为双侧对称性中高频下降的重度至极重度感音神经性听力下降,典型听力图表现为特征性的‘U’型或陡降型。4例为语后聋,1例语前聋患儿未能通过新生儿听力筛查。根据系谱图分析,该家系均为男性患病,双亲正常,符合X连锁隐性遗传模式,同时先证者耳聋基因筛查亦为阴性。结论本家系的临床听力学及遗传学特征分析符合X连锁隐性遗传,进一步将通过外显子测序探索该家系耳聋致病基因。
家系;X连锁隐性遗传;表型;遗传性耳聋
Acknow ledgment:We thank the patients and the other participantswho took part in this study for their collaboration.by the NationalNature Science Foundation of China(Grant No.81170923,81470705,81300833),Thiswork was supported by the National Basic Research Program of China(Grant No.2014CB541702,2014CB943003),,by the Science and Technology Projects in Hunan Province(GrantNo.13JJ4023)
Theauthorsdeclare that they haveno competing interests.
耳聋是最为常见的出生缺陷和感音神经性疾病[1],超过50%的语前聋为遗传性因素引起,70%遗传性耳聋均为非综合征型耳聋[2](nonsydromic hearing loss,NSHL),其中NSHL中常染色体隐性遗传(DFNB)约占77%,常染色体显性遗传(DF⁃NA)约占22%,X连锁遗传仅占1%~2%,线粒体遗传约占1%。综合征型遗传性耳聋中X连锁遗传报道较多[3],而临床X连锁非综合征型遗传性耳聋非常少见,根据目前X连锁耳聋基因报道可知,该类遗传性耳聋的起病年龄和听力损失程度呈现极大的异质性。王秋菊等[4-5]报道了一个Y连锁遗传的中国耳聋大家系和X连锁隐性遗传聋哑家系。
本研究报道一个湖南省X连锁非综合征型耳聋家系,针对其听力学特征及遗传性特征进行分析,并针对该家系制定耳聋致病基因的研究策略,现报道如下。
1 资料和方法
1.1家系资料的采集
本研究家系资料的调查研究工作获得湘雅医院伦理委员会认可,由湘雅医院耳鼻咽喉头颈外科家系采集小组完成。家系位于湖南省某市,先证者为1例7岁双耳对称性、中度感应神经性耳聋的男性患者,家系信息采集小组对该先证者及其各亲属成员进行了家系调查,所有家系成员均签署知情同意书,对家系中的21名成员完成问卷式调查资料、听力学检查、粗略智力评估、简易电耳镜检查、体格检查(包括专科检查和全身常规检查)等检查,EDTA采血管采集外周静脉血3~8m l,提取基因组DNA并保存。
1.2遗传方式判断标准
目前国内外研究者比较认可和通用的分类方法是遗传性耳聋分为综合征型耳聋(SHL)和非综合征型耳聋(NSHL)。SHL除了耳聋症状外,尚伴有眼、肾、甲状腺、骨等其他器官发育异常或功能障碍。NSHL根据遗传方式可分为常染色体隐性遗传、常染色体显性遗传、X染色体连锁遗传和线粒体母系遗传,不同的遗传方式可呈现不同的遗传学特征及临床特征,控制一种性状或遗传病的基因位于性染色体上,该基因必将随着性染色体的传递而传递,其遗传方式称为性连锁遗传(sex-linked inheri⁃tance)或称伴性遗传。根据人类的性染色体不同以及性染色体上致病基因的性质不同,可将性连锁遗传分为X连锁显性遗传、X连锁隐性遗传和Y连锁遗传,X连锁遗传性耳聋具有特征性的交叉遗传现象。
1.3听力学检测及标准
本部分工作均由听力学专科技师以及临床专科医师依照规范标准完成。运用丹麦Madsen502便携式听力计对参与本研究的家系成员进行常规听力学检测,包括纯音测听、声导抗,先证者在湘雅医院耳鼻咽喉头颈外科学进行了听性脑干诱发电位(ABR)检测,根据WHO预防聋和听力损失项目报告(1997年,日内瓦),按较好耳0.5Hz~4KHz四个频率计算平均听阈,听力损失程度分级[4]:轻度(26~40dBHL);中度(41~60dBHL);重度(61~80dB⁃HL);极重度(>81dBHL)。根据听力损失的频率特征分为:低频<=0.5kHz;中频>0.5kHz<=2kHz;高频>2kHz<=8KHz;超高频>8KHz。
1.4耳聋基因筛查
对于耳聋基因定位克隆研究工作的研究方法往往灵活多变,利用Sanger测序对先证者进行线粒体基因全序列、GJB2以及GJB3进行筛查,排除常见已知致病突变位点。
2 结果
2.1家系基本资料
该4代家系居住于湖南省某市,现存家系成员28人(16男,12女),其中确诊患者5人,均为男性,耳聋患者年龄最大者45岁,最小者0岁,均以听力损失为单一症状,无噪声接触史,除Ⅲ:5之外,均无耳毒性药物用药史,2例伴耳鸣症状,均无前庭功能症状,体格检查未见明显器官、系统异常,其中4例自幼出现听力损失,但言语表达清晰,1例语前聋,未通过新生儿听力筛查。先证者为Ⅳ-1,男,7岁,因双耳听力下降就诊于湘雅医院耳鼻咽喉头颈外科学门诊,纯音测听示双耳、对称性中度感音神经性聋,无眩晕史,顺产,无肾病及眼科病史。双耳听力曲线均呈‘U’型;双耳声导抗A型曲线;颞骨HRCT未见异常,排除中耳及内耳解剖病变。家系内成员均生活在同一环境,排除非遗传性因素对耳聋患者听力损失的影响。家系成员资料详见表1。
2.2家系听力学特征及遗传性特点
2.2.1家系听力学特征对家系内成员进行纯音听阈检测(部分成员听力曲线见图1),该家系第二代直系血亲成员3人,明确耳聋患者2人,Ⅱ:5自幼耳聋,进展迅速,表现为极重度耳聋,听力曲线呈陡降型,Ⅱ:1成员发病2年,进展缓慢,表现为双侧对称性、轻度耳聋,平坦型听力曲线,双耳声导抗A型图,双耳DPOAE未通过,ABR左耳气导阈值40dBnHL,右耳30dBnHL,因X连锁遗传异质性,仍将其视为携带者;第Ш代中有血缘关系的成员共8人,明确耳聋患者2人,Ⅲ:5和Ⅲ:9均自幼发病,迅速进展为重度~极重度耳聋,且Ⅲ5言语欠清晰,可能是语前聋或语中期聋;第Ⅳ代有血缘关系的成员共7人,明确耳聋患者2人,先证者Ⅳ:1表现为双侧对称性、中频为主的中度听力损失,7岁开始发病,言语发育正常,双耳声导抗A型图;ABR双侧气导阈值60dBnHL,颞骨HRCT未见明显异常(图2),Ⅳ:3出生未通过听力筛查,复查ABR双耳80dBnHL可诱出V波,左耳气导反应阈值50dBnHL,右耳80dBnHL,提示重度耳聋,头颅MRI未见明显异常。
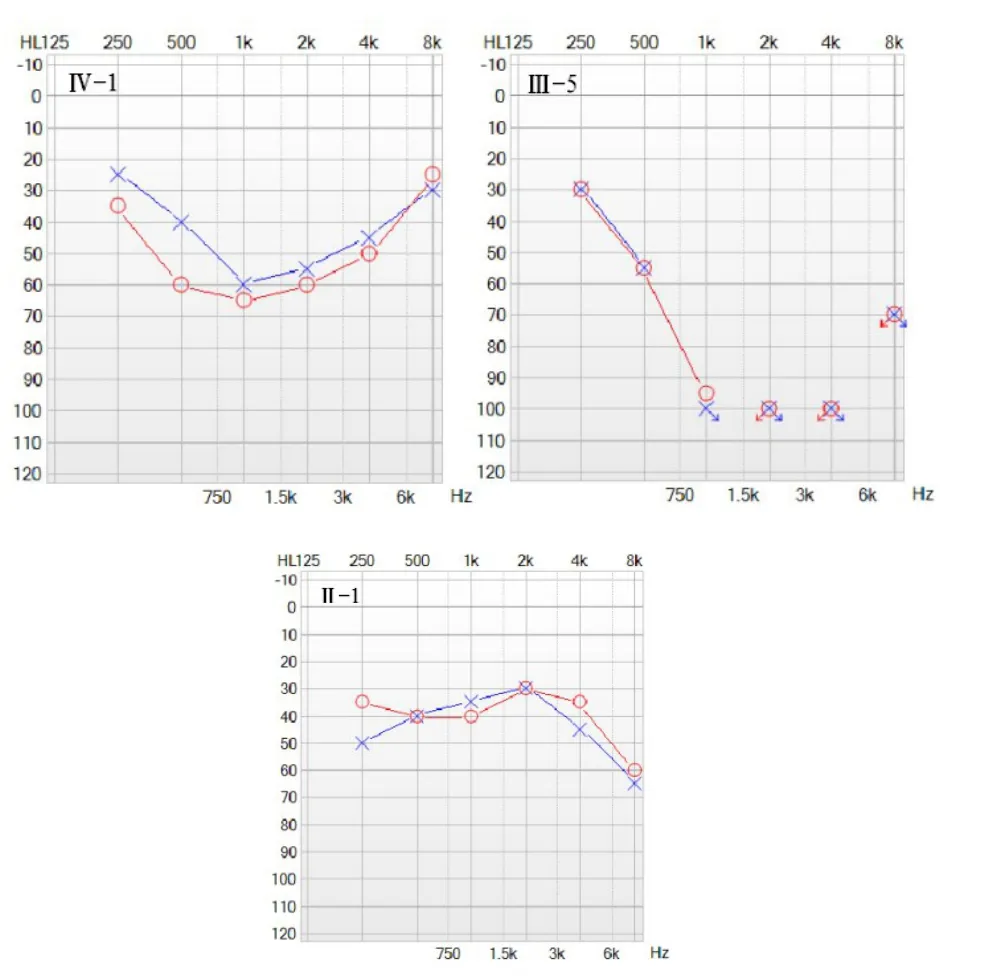
图1 部分家系成员听力图
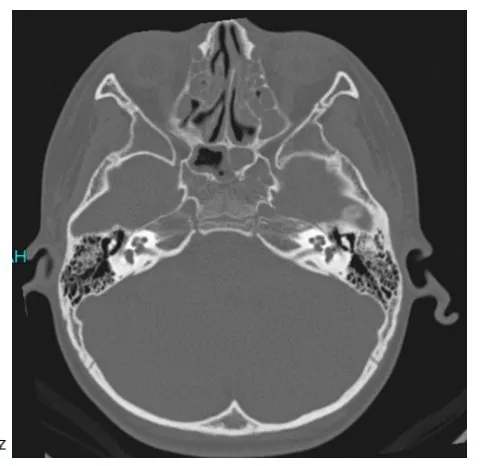
图2 Ⅳ:1颞骨HRCT
2.2.2家系的遗传学特征
根据本研究家系调查小组采集到的信息资料和整理分析,应用cyrillic3.0软件绘制系谱图(见图3),通过系谱图可知,家系共4代,现存3代成员28人,分析本家系遗传系谱,可发现1.家系中患者几乎都是男性;2.双亲无病时,儿子50%可能患病,女儿不发病,携带者母亲的致病基因可以遗传给外孙;3.男性患者的子女都是正常,且各代男性患者为表兄弟、舅外孙等关系,家系遗传图谱呈现不连续遗传的现象,以上特征符合X连锁隐形遗传模式。

图3 耳聋家系系谱图Fig.3 Pedigree of the Chinese fam ily w ith X-linked Heriditary NSHL
2.3耳聋基因筛查结果
该家系先证者耳聋基因筛查GJB3:c.-557_-556insC为已报道多态,GJB2未发现突变,线粒体全基因序列Sanger测序共发现42个多态位点。
3讨论
绝大多数非综合征型遗传性耳聋均由常染色基因引起,X连锁相关大约占1%~2%,目前明确的X连锁基因座位共4个,并已广泛采用新的命名,分别为DFNX1(PRPS1)、DFNX2(POU3F4)、DFNX4(SMPX)、DFNX6(COL4A6),其致病基因已先后被克隆(http://hereditaryhearingloss.org)。
DFNX1:PRPS1,原DFN2位点,Tyson(1996)通过一个四代英-美家系定位在Xq22,家系内男性患者先天性极重度感音神经性聋,女性携带者为轻-中度耳聋,高频损失为主。Manolis[6]在一个美国家系中报道,男性患者表现为低中频损失为主的上升型曲线,语后聋,渐进性加重,发病年龄在7岁~20岁之间,家系内女性携带者听力损失较轻。Cui(2004)[7]在一个大的X连锁语后感音神经性聋中国家系中也定位在DFN2位点。Liu XZ etal.(2011)[8]报道在一个大的语后非综合征型中国耳聋家系,通过连锁定位于同DFN2重叠区内5.41cM大小区间,该X连锁隐形遗传性耳聋家系中男性表现对称性、渐进性加重,24~50岁之后进展为重度-极重度耳聋,听力曲线表现平坦型,一例52岁女性携带者听力正常,部分女性携带者(47岁~64岁)表现单侧或不对称性轻度-重度耳聋,还有一例71岁女性表现为单侧重度-极重度耳聋。通过区间内候选基因筛查发现PRPS1(MIM 300401)基因在家系中共分离,结合之前报道的3个家系共发现了四个错义突变(D65N,A87T,I290T,以及G306R)。PRPS1基因突变可导致磷酸核糖焦磷酸合成酶1(PRS1)活性异常,PRS1属于一个体内催化ATP和5-磷酸核糖合成磷酸核糖焦磷酸(PRPP)激酶家族,PRPP是体内嘌呤、嘧啶以及嘧啶核苷酸从头合成和补救合成的重要底物,也是该通路上的重要调节因子,该酶活性的异常改变,导致嘌呤和嘧啶代谢的紊乱,从而导致耳聋的发生。Liu XZetal.(2011)报道的四个点突变,预测并未导致PRS1蛋白重大结构性改变,分子模型预测突变影响酶活性中心—ATP结合域,以家系内正常男性成员以及无血缘关系正常人对照,在男性患者的红细胞和皮肤成纤维细胞中,检测到PRS1活性降低44%~45%,相较两类综合征型遗传性耳聋,X性连锁Charcot-MarieTooth氏综合征5型(CMTX5)(62%)和Arts综合征(>92%),PRS1活性降低程度相对较低,故表现为非综合征型耳聋表型。为了维持机体内生化平衡,其活性必须维持在一个相对适当的范围[9]。

表1 家系患者临床表现Table.1 Clinical featuresof the Chinese fam ily w ith X-linked Heriditary NSHL
DFNX2:POU3F4,原DFN3位点,X连锁遗传性耳聋中检出率最高的基因,定位于Xq21.1[10],de Kok(1995)[11]在14个不相关的X连锁遗传性耳聋患者和50个不同种族的女性对照样本中,检测到3个无义突变和2个错义突变,DFNX2耳聋患者可表现特征性的混合型耳聋,也可表现为因镫骨固定引起的传导性耳聋以及渐进性加重的感音神经性聋,有时极重度感音神经性聋可能掩盖传导性损失部分,伴或不伴前庭功能紊乱,家系中男性患者语前发病,女性携带者可不发病或症状轻微,遗传图谱呈现明显“隔代遗传”规律。患者CT往往表现内听道异常膨大,同时内听道和内耳之间通道扩大,其结果是导致外淋巴液压力增高,出现镫骨手术中的“井喷”现象,王秋菊等[12]报道了一个中国江西五代耳聋大家系,其中8例男性患者表现为先天性极重度感音神经性聋,且颞骨高分辨率CT均表现出内听道外侧端异常扩大,女性携带者听力正常,呈现X连锁隐形遗传性方式。该基因突变引起的遗传性耳聋在美、欧、亚均有报道,影响广泛[17]。
DFNX4:SMPX,原DFN6位点,由Castillo (1996)[13]等通过一个西班牙5代X连锁非综合征型耳聋家系的连锁分析,定位在Xp22,家系内男性患者为语后聋,最初高频损失为主,随着年龄逐渐加重,成年后表现为对称性、累及全频的重度-极重度感音神经性聋,发病年龄5~7岁。女性携带者不完全外显,最早30岁发病,一般40岁左右发病,表现为对称性、高频为主的中度感音神经性聋,患者均无前庭功能异常以及耳鸣。2011年Huebner在不同的X连锁NSHL家系中克隆了SMPX基因,并预测该基因同内耳发育和维持内耳毛细胞机械应力密切相关。Schraders等[14]报道的芬兰家系中男性患者发病年龄在2~10岁,并可在20岁以前迅速进展为极重度耳聋,女性患者在3~48岁发病,且个体间表型差异很大,甚至双耳之间存在差异。Hueb⁃ner等[15]报道的德国语后聋家系同Schraders报道的家系很相似,男性患者3~7岁发病,女性患者发病年龄在20~30岁,男女性听力损失特征及进展情况相近,亦无前庭功能障碍以及耳鸣。SMPX基因编码一种小的细胞骨架相关肌肉蛋白,88个氨基酸,但是小鼠内耳免疫荧光定位显示Smpx主要位于支持细胞,在毛细胞中低表达,SMPX基因功能域尚不清楚,其致病机制有待进一步验证。
DFNX6:Rost等(2014)[16]通过一个3代匈牙利X连锁非综合征型聋家系,通过二代测序克隆了COL4A6基因,该家系中只有男性出现先天性重度耳聋,而女性患者(9~81岁)表现正常或仅仅轻度-中度听力损失,且男性患者颞骨HRCT和MRI显示耳蜗发育畸形。COL4A6基因共45个外显子,编码Ⅳ型胶原α-6链,可同COL4A5基因编码的两条α-5链可形成异源三聚体,两个基因的突变可以导致肾病、耳聋、白内障综合征,即Alport综合征。
本研究家系中,第二至第四代均有患者,由于成员Ⅰ-1已去世,其发病情况亦无法追溯,成员Ⅲ-5的母亲Ⅱ-1表现轻度耳聋,其余Ⅳ:1/Ⅳ:3两名男性患者的母亲均无耳聋表型,考虑X连锁非综合征型遗传性耳聋女性携带者可以出现轻度听力损失,往往成年后发病,但发病年龄异质性显著,其父亲Ⅰ-2无耳聋症状,故Ⅱ-1是致病基因携带者。Ⅱ-3和Ⅲ-1成员右耳听力正常,左耳均表现为轻度听力损失,Ⅲ-3成员听力检测虽正常,但考虑其尚未到发病年龄,符合X连锁遗传性耳聋家系女性携带者表型异质性,而此三名成员的子代明确有耳聋患者,故Ⅱ-3、Ⅲ-1、Ⅲ-3成员是耳聋基因携带者。家系中男性患者有先天性耳聋,也有语后聋,但起病年龄均较早,双侧对称性加重,20岁左右发展为中高频损失为主的重度-极重度感音神经性聋,其中成员Ⅲ-5比较特殊,需要重点鉴别,经过反复询问及核对,明确其3岁时有庆大霉素用药史(具体剂量不详),但用药后没有立即出现听力下降等症状,而是7~8岁出现听力下降,为排除其药物性聋可能,线粒体DNA筛查未发现已知耳聋突变位点(未提供),根据其听力损失特征,与家系中其他男性患者的耳聋表型比较分析,Ⅲ-5的听力损失更可能是遗传性因素导致,故暂时仍将其视为家系内患者。
按照传统耳聋基因定位克隆策略,需先行筛查以上四个基因,但COL4A6、SMPX、POU3F4、PRPS1,该4个基因外显子比较大,尤其COL4A6(45个外显子),候选逐个筛查效率较低。鉴于家系先证者线粒体全序列筛查共发现42个多态位点,初步排除母系遗传可能性。GJB2/GJB3筛查亦未发现致病突变。近年发展起来的新一代高通量测序技术(Next-generation sequencing,NGS),目前广泛应用于孟德尔遗传病的致病基因的鉴定研究,近5年得益于该项技术的帮助,超过10个新的耳聋基因被鉴定,充分展现了该技术的高效性、快速性以及应用前景,本课题组拟选取表型明确的一例患者Ⅳ-1进行全外显子组高通量测序,初步排除已知耳聋基因之后,补送Ⅳ-3和Ⅲ-9两例患者以期找到该家系的耳聋致病基因。
1 HILGERTN,SMITH RJ,Van CAMPG.Forty-Six GenesCausing Nonsyndromic Hearing Impairment:Which Ones Should be Ana⁃lyzed in Dna Diagnostics?MutatRes,2009:681(2-3):189-196.
2 Van CAMPG,WILLEMSPJ,SMITH RJ.Nonsyndromic Hearing Impairment:Unparalleled Heterogeneity.Am JHum Genet,1997: 60(4):758-764.
3 SMITH R,SHEARER AE,HILDEBRAND MS,et al.Deafness and Hereditary Hearing LossOverview.GeneReviews,2008.
4王秋菊,杨伟炎,韩东一,等.Y-连锁遗传性耳聋:中国一大家系的听力学表型特征.中华耳科学杂志.2004(02):4-10.
QiujuWang,Weiyan Yang,DongyiHan,etal.The Audiologic as⁃pects in Chinese Y-linked hereditary hearing impairment pedi⁃ gree.Chinese JournalofOtology,2004(02):4-10.
5王秋菊,杨伟炎,吴子明,等.X连锁隐性遗传聋哑(Deaf-Mute)家系的遗传学特征分析.遗传.2004(05):579-583.
Qiuju Wang,Weiyan Yang,ZimingWu,et al.Genetic Analysis in a Chinese Deaf-mute Family with X linked Recessive Inheri⁃tance.Hereditas,2004(05):579-583.
6 MANOLISEN,EAVEYRD,SANGWATANAROJS,etal.Heredi⁃tary Postlingual Sensorineural Hearing Loss Mapping to Chromo⁃some Xq21.Am JOtol,1999:20(5):621-626.
7 CUI B,ZHANG H,LU Y,et al.Refinement of the Locus for Non-Syndromic Sensorineural Deafness(Dfn2).JGenet,2004:83 (1):35-38.
8 LIU X,HAN D,LI J,et al.Loss-of-Function Mutations in the Prps1Gene Cause a Type ofNonsyndromic X-Linked Sensorineu⁃ral Deafness,Dfn2.The American Journal of Human Genetics, 2010:86(1):65-71.
9李建忠,袁慧军,韩东一.Prps1基因突变与遗传性耳聋.中华耳科学杂志.2010,8(1):57-62.
Jianzhong Li,Huijun Yuan,Dongyi Han.The Mutation of Prps1 and Hereditary hearing loss.Chinese Journal of Otology,2010,8 (1):57-62.
10 WALLISC,BALLO R,WALLISG,et al.X-Linked Mixed Deaf⁃nesswith Stapes Fixation in a Mauritian Kindred:Linkage to Xq Probe Pdp34.Genomics,1988:3(4):299-301.
11 de KOK YJ,van der MAAREL SM,BITNER-GLINDZICZM,et al.Association Between X-Linked Mixed Deafness and Mutations in the Pou Domain Gene Pou3F4.Science,1995:267(5198): 685-688.
12 WANG Q,LI Q,RAO S,et al.A Novel Mutation of Pou3F4 Causes Congenital Profound Sensorineural Hearing Loss in a Large Chinese Family.The Laryngoscope,2006944-950.
13 DEL CI,VILLAMAR M,SARDUY M,et al.A Novel Locus for Non-Syndromic Sensorineural Deafness(Dfn6)Maps to Chromo⁃some Xp22.Hum MolGenet,1996:5(9):1383-1387.
14 SCHRADERSM,HAASSA,WEEGERINKNJD,etal.Next-Gen⁃eration Sequencing Identifies Mutations of Smpx,Which Encodes the Small Muscle Protein,X-Linked,as a Cause of Progressive Hearing Impairment.The American Journal of Human Genetics, 2011:88(5):628-634.
15 HUEBNERAK,GANDIAM,FROMMOLTP,etal.NonsenseMu⁃tations in Smpx,Encoding a Protein Responsive to Physical Force, Result in X-Chromosomal Hearing Loss.The American Journalof Human Genetics,2011:88(5):621-627.
16 ROST S,BACH E,NEUNER C,et al.Novel Form of X-Linked Nonsyndromic Hearing Loss with Cochlear Malformation Caused by a Mutation in the Type Iv Collagen Gene Col4a6.Eur JHum Genet,2014:22(2):208-215.
17黄邦清,曾佳玲,苏钰,等.一个X连锁隐性遗传耳聋基因POU3F4的新突变.中华耳科学杂志.2014(01):57-60.
Bangqing Huan,Jialing Zeng,Yu Su,etal.A novel POU3F4 gene mutaion for X-linked recessive hereditary hearing loss.Chinese JournalofOtology,2014(01):57-60.
Clinicaland genetic featuresof a Chinese fam ilyw ith nonsydrom ic X-linked recessivehearing loss
Niu Zhijie1,2,Feng Yong1,2,3,Mei Lingyun1,2,Sun Jie1,2,4,Cheng Hongsheng1,2,HeChufeng1,2,Liu Yalan1,2, Wang Xueping1,2,Wen Jie1,2,Jiang Lu1,2
1DepartmentofOtolaryngology-Head and Neck Surgery,Xiangya Hospital,CentralSouth University, Changsha,410008,China
2 Key Laboratory ofOtolaryngologyMajor DiseaseResearch ofHunan Province,Changsha,410008,China
3StateKey Laboratory ofMedicalGenetics,Central South University,Changsha,410078,China
4DepartmentofOtorhinolaryngology,FirstAffiliated HospitalofXinjiang MedicalUniversity,Urumqi,830011,China
Jiang Lu Email:Lqjtx@163.com
Objective To report the clinical and genetic characteristics of a large Chinese pedigreew ith X-linked recessive non-syndromic hearing loss.Methods We used deafness-questionnaires to collect detailedmedicalhistory information.Syndromic hearing losswas ruled outvia clinicalexam ination,otoscopy and pure-tone audiometry.We plotted the pedigree based on the genetic and audiology characteristics of this fam ily and screened GJB2,GJB3 andm tDNA to excludewell known pathogenicmutations.Results A total of 28memberswere alive in this four-generations fam ily, and 5males were found to be hearing-impaired.Most of the patients had moderate to profound sensorineural hearingloss affecting predominantly themiddle and high frequencies.One child w ith apparently pre-lingual hearing loss failed the newborn hearing screening.One carrier female showed m ild hearing loss.The characteristic audiometric configuration was either a U or a steep sloping pattern.We did not find any causativemutations by screening the three common deafness genes.Conclusions Pedigree analysisof this fam ily indicatesan X-recessive inheritance pattern of hearing impairment,in which affected-malemembers showed pre-lingualor post-lingual,symmetricaland fast-progressing hearing loss.Whole-exome sequencing is probably needed to identify the disease-causing gene in this fam ily.
Pedigree;X-linked hearing loss;Phenotype;Hereditary deafness;
R764
A
1672-2922(2017)02-195-6
2015-12-17审核人:冰丹)
10.3969/j.issn.1672-2922.2017.02.011
国家自然科学基金项目(GrantNo.81300833,81170923,81470705)、国家重大科学研究计划项目(GrantNo.2014CB541702,2014CB943003)及湖南省自然科学基金(GrantNo.13JJ4023)共同资助。
牛志杰,男,湖南人,在读博士研究生,专业方向:遗传性聋为主的耳科学基础与临床研究。
蒋璐,Email:Lqjtx@163.com
猜你喜欢
中国现代医生(2022年19期)2022-11-04
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中国肿瘤临床(2022年14期)2022-08-09
昆明医科大学学报(2022年4期)2022-05-23
中国听力语言康复科学杂志(2021年6期)2021-12-21
中医眼耳鼻喉杂志(2021年1期)2021-07-22
中国生殖健康(2020年4期)2021-01-18
家庭百事通·健康一点通(2019年8期)2019-08-29
中南林业科技大学学报(2019年4期)2019-04-08
