
苹果Hsf家族成员的序列特征、表达与进化分析
2017-06-28 12:36张国俊王婷婷胡利宗李书粉高武军
华北农学报 2017年2期
张国俊,王婷婷,胡利宗,李书粉,高武军
(1.新乡医学院 基础医学院,河南 新乡 453003;2.河南师范大学 生命科学学院,河南 新乡 453007;3.漯河职业技术学院 食品工程系,河南 漯河 462000)
苹果Hsf家族成员的序列特征、表达与进化分析
张国俊1,2,王婷婷3,胡利宗2,李书粉2,高武军2
(1.新乡医学院 基础医学院,河南 新乡 453003;2.河南师范大学 生命科学学院,河南 新乡 453007;3.漯河职业技术学院 食品工程系,河南 漯河 462000)
为全面了解苹果基因组中热激转录因子(Hsf)的序列特征及进化,采用生物信息学手段,在苹果全基因组水平上鉴定出50个MdHsf基因,并对其系统发育关系、序列特征、表达情况以及选择压力进行详细分析。系统发育与序列分析显示:与拟南芥和水稻相似,50个MdHsf基因可分为A、B、C 3个亚族;2个或多个MdHsf基因位于同一个末端进化支,说明该基因家族在苹果中发生了物种特异性扩增;尽管MdHsf基因的内含子数目和长度变异较大,但其蛋白的保守基序和功能结构域具有较高的保守性,这可能与功能约束有关。基于EST数目,可推知:除了MdHsfA2a和MdHsfA3a/b/c等14个基因没有相应的EST外,其余72%的基因都有转录活性。选择压力检测和结构建模分析显示:在36个MdHsf蛋白的选择压力检测中,位点模型未鉴定到正选择位点的存在;而在显著水平下(P<0.05),分支-位点模型在d和e进化分支上,共检测到5个正选择位点,它们是28R、30L、35D、51M、67V,其中28R和30L位于Hsf结构域中,35D、51M和67V位于Hsf结构域之外,这说明除了MdHsfA4d/e和MdHsfC1a/b发生快速进化外,其他成员受控于纯净选择,具有高度保守性。综合以上研究结果,苹果基因组中存在多种热激转录因子,其蛋白的保守基序和功能结构域具有较高保守性,大多具有转录活性,在进化上该家族受纯净选择主导。
苹果;热激转录因子;表达;进化
热激转录因子(Heat shock transcription factor,Hsf)是一种反式作用因子,能与热激元件相互作用,通过调控Hsp基因(Heat shock protein,Hsp)的表达,参与生物体的热应激反应[1]。由于热激蛋白基因的表达受到热激转录因子的调控,因此,Hsf在植物抗击热胁迫反应中扮演着十分重要的角色[2]。基于足迹法和亲和层析法,Wiederrecht等[3]于1988年首次在酵母(Saccharomycescerevisiae)中克隆得到第一个Hsf基因,随后,有关Hsf基因的克隆与研究工作主要集中于少数模式动物,例如果蝇(Drosophilamelanogaster)[4]、小鼠(Musmusculus)[5]和人类(Homosapiens)[6]等。几乎同时,Scharf等[7]以酵母Hsf的保守结构域为检索序列,在番茄(Solanumlycopersicum)中克隆到了3个Hsf基因。此后,研究人员陆续在拟南芥(Arabidopsisthaliana)[8]和水稻(Oryzasativa)[9]等物种中,克隆并鉴定了多个Hsf基因。动物、植物和微生物Hsf基因数目的比较分析表明,植物Hsf基因不但成员数目多,而且具有功能的冗余性和多样性等特点。与动物相比,由于固着生长的植物面对更为复杂的逆境胁迫,相应地需要更多功能多样化的防御体系来维持生理稳态,可推测植物很可能具有更多的热激转录因子成员参与热激反应。因此,在基因组水平上,鉴定所有Hsf基因成员仍然是一项重要而艰巨的任务。
近年来,随着测序技术的快速发展,越来越多的模式植物全基因组测序工作已经完成,这为鉴定与分析Hsf基因家族提供了便捷。在全基因组水平上,许多植物Hsf基因家族包括多个成员,每个成员都含有保守的DNA结合结构域,例如拟南芥、水稻、玉米(Zeamays)、高粱(Sorghumbicolor)、大豆、杨树(Populustrichocarpa)、番茄、大白菜(Brassicarapassp.pekinensis)、胡萝卜(Daucuscarota)和茶树(Camelliasinensis)中分别至少有21,25,25,24,52,27,24,35,35,16个Hsf基因[10-16]。根据蛋白序列、结构和进化上的关系,植物中Hsf蛋白家族存在A、B、C共3类成员。其中,A类Hsf主要负责热激基因表达的调控;B类Hsf虽然具有DNA结合活性但却没有热激诱导的转录激活活性,可能与A类Hsf共同发挥作用,C类Hsf的作用尚不清楚[17-18]。
目前,对Hsf基因的研究主要集中在模式植物上,而对果树的相关研究报道还比较少。苹果(Malusdomestica)全基因组测序的完成为系统剖析MdHsf基因家族提供了便捷[19]。2012年,Giorno等[20]在苹果基因组中鉴定了25个Hsfs,并进行了分类和表达分析。分析发现,在苹果基因组中存在着更多的Hsfs。为此,本研究围绕基因结构、保守基序、功能结构域、蛋白三维结构、表达与快速进化等问题,对苹果MdHsf家族基因的50个成员进行了全面而系统的分析,以期为克隆和鉴定苹果MdHsf的生物学功能奠定基础,为植物Hsf基因的系统发育关系与快速进化机制提供线索。
1 材料和方法
1.1 苹果Hsf基因的鉴定和进化树构建
根据已有报道,拟南芥和水稻Hsf基因家族成员的相关信息直接查文献获得[10-11],其相关序列主要来源于3个数据库:Phytozome v8.0(http://www.phytozome.net/)、JGI(http://genome.jgi.doe.gov/programs/plants/index.jsf)和NCBI(http://www.ncbi.nlm.nih.gov/)。为获取苹果Hsf基因,本研究分别以拟南芥Hsf基因、水稻Hsf基因和植物Hsf结构域(PF00447)的一致性序列为检索序列,对苹果基因组数据库进行了Blast搜索(E=0.01)。移走冗余序列,利用Pfam工具(http://pfam.sanger.ac.uk/)对所得Hsf蛋白序列进行分析,若存在Hsf结构域(PF00447),则认为该蛋白质属于Hsf家族成员。如果同一个基因座有多个转录本,选择最长的转录本作为代表,每个基因座只算作1个基因成员。为阐明苹果Hsf基因的进化关系,构建了拟南芥、水稻和苹果Hsf蛋白的进化树。具体步骤如下:在默认参数下,利用MUSCLE软件对Hsf蛋白序列进行多重比对分析[21];基于比对结果,采用极大似然法(Maximum likelihood method)构建系统发育树,进化树的构建和输出均由MEGA软件完成[22]。
1.2 苹果Hsf基因的序列特征分析
通过Hsf基因的DNA和cDNA序列的比较,可确定Hsf基因的结构,其结构模式图由GSDS软件(http://gsds.cbi.pku.edu.cn/)绘制。利用MEME工具(http://meme.sdsc.edu/)对苹果Hsf蛋白的保守基序进行分析。参数设置如下:同一基序在一条序列中出现的次数为0或者1,基序长度为6~200个氨基酸残基,基序最大发现数目10个,其他参数为默认值。此外,利用Pfam工具(http://pfam.sanger.ac.uk/)对所有Hsf蛋白的功能结构域进行鉴定,并对结构域的排列方式进行分析。
1.3 苹果Hsf基因的表达分析
不同组织来源EST数目的统计分析不仅能推测基因的转录活性,而且能反映基因的表达水平。以苹果Hsf基因的编码序列为检索序列,利用Blast工具在GenBank中dbEST数据库搜索相应的EST序列,参数设置为默认值。基于最佳匹配的EST序列,链接到UniGene,利用该UniGene的EST组织表达谱推测Hsf基因的表达情况。
1.4 苹果Hsf基因选择压力的检测
由于50个苹果Hsf基因间的差异度较大,不便于进行选择压力研究,因此,有必要对这些基因进行分组。首先,利用ClustalX软件对Hsf基因进行多重序列比对[23]。然后,根据3个标准进行分组:①氨基酸水平上组内的平均相似度大于30%;②组内每个成员氨基酸序列长度大于总长度的50%;③每个组内的成员数目大于3个基因。上述标准用在线工具BLASTclust(http://toolkit.tuebingen.mpg.de/blastclust)计算检测,其中36个苹果Hsf基因符合上述标准。最后,利用Gblock软件[24]移走这些基因多序列比对中的高度分歧区,对剩余的同源区进行进一步的选择压力分析。36个苹果Hsf基因同源区的密码子比对文件由Pal2nal工具[25]生成,其树文件由TreeView软件[26]产生。利用PAML3.15(http://abacus.gene.ucl.ac.uk/software/paml.html)软件包中的CODEML程序对苹果Hsf基因的选择压力进行分析,其中,位点特异模型用于每个组内的选择压力检测,而位点分支模型用于每个分支的选择压力检测[27]。为将MdHsfA4d、MdHsfA4e、MdHsfC1a和MdHsfC1b蛋白的正选择位点定位在三维结构中,本研究基于同源建模方法,利用SWISS-MODEL服务器对4个苹果Hsf蛋白的三维结构进行预测[28],其中MdHsfA4d、MdHsfC1a和MdHsfC1b以人类Hsf1蛋白(2lduA)为模板,而MdHsfA4e以乳酸克鲁维酵母Hsf蛋白(3hsfA)为模板。同时,经Loop区优化、能量最小化和动力学模拟等步骤,最后得到最佳三维构象。
2 结果与分析
2.1 苹果Hsf基因的鉴定及其系统发育分析
分别以AtHsf、OsHsf和植物Hsf结构域(PF00447)的一致性序列为检索序列,利用Blast同源搜索方法在苹果基因组数据库中挖掘Hsf候选基因,移走冗余序列后,利用Pfam对所有蛋白进行特征结构域扫描。最终,在苹果基因组中共鉴定了50个Hsf基因,其中包括前人鉴定得到的23个Hsf基因。表1列举了这些基因的名称、类型、登录号、叠连群与染色体位置。
为阐明不同植物Hsf基因的进化关系,对50个MdHsf以及具有代表性的21个AtHsf和25个OsHsf的蛋白序列进行亲缘关系分析,获得环状进化树(图1)。其中,苹果和拟南芥隶属于双子叶植物,而水稻属于单子叶植物。结果表明:3种代表性植物的96个Hsf蛋白可分为3个亚家族,即A、B、C亚家族。根据MdHsf与AtHsf蛋白的系统发育关系,并参考拟南芥该家族的名称,对每个MdHsf基因进行了编号和命名(表1、图1)。从亚家族的成员数目看,苹果的亚家族A包括37个基因,亚家族B包括11个基因,亚家族C仅包括2个基因。其中,23个基因为Giorno等[21]已经鉴定过的(表1)。通过对比该分类结果和Giorno等的分类结果,对这23个基因的分类是完全一致的。从亲缘关系角度看,绝大多数苹果Hsf蛋白优先与拟南芥同源基因聚在一起,然后再与水稻相应同源基因聚为一簇,这与物种进化关系具有较高一致性(图1)。
2.2 苹果Hsf基因的序列与进化特征
本研究基于苹果Hsf蛋白全长序列构建了该家族的进化树(图2-A),并对苹果50个MdHsf基因的结构进行分析(图2-B)。结果显示,除了MdHSFA9g基因没有内含子外,其余的MdHsf基因都至少有1个内含子。若以内含子数目为准,具有内含子的49个苹果MdHsf基因的结构可分为6种类型,它们分别有1,2,3,4,6,10个内含子。不同类型的基因结构具有不同的频率:1个内含子类型基因数目最多,包括MdHsfA6c等19个基因;2个内含子类型基因数目次之,包括MdHsfA3c等13个基因;3个内含子类型包括MdHsfA6e等10个基因;4个内含子类型包括Mdhsfa6a等5个基因;而6个内含子和10个内含子类型数目最少,仅各包含MdHsfAqe和MdHsfAbe(图2-B).
注:Md.苹果;Hsf.热激转录因子;Chr.染色体。
Note:Md.Apple;Hsf.Heat shock factor;Chr. Chromosome.
其次,蛋白结构域分析显示,绝大多数苹果Hsf仅仅包括Hsf结构域。但MdHsfA6e、MdHsfA8a/b、MdHsfB1a、MdHsfA10b和MdHsfA10e不但含有典型Hsf结构域,而且还包括额外的功能结构域,例如MdHsfA6e包括1个ARD,MdHsfA8a/b各包括1个EF,MdHsfB1a包括RCC,MdHsfA10b包括4个串联的HPR,MdHsfA10e包括2个串联的WD结构域(图2-C)。利用MEME软件对50个苹果Hsf蛋白保守基序进行预测,结果显示,在该蛋白家族中共检测到10个保守基序,依次编号为1~10(图2-D)。在苹果Hsf蛋白的A亚家族中,除了MdHsfA9g、MdHsfA9b、MdHsfA10d和MdHsfA10c只包含一个保守基序外,其他的蛋白都至少具有2个保守基序,其中包括MdHsfA6d等在内的23个蛋白都至少包含5个保守基序,这些保守序列的组成和排列顺序具有较高的保守性。与A亚家族保守基序相比,苹果Hsf蛋白的B亚家族蛋白最多仅包含5个保守基序,并且不包括保守基序3,5,6,7和8。C亚家族的保守基序模式与A亚家族十分相似,但该亚家族并不包括保守基序5,6,7和8,因此,可推测保守基序5,6,7和8是A亚家族所特有的。
为了解亲缘关系比较近的苹果Hsf基因是否具有相同或相似的序列特征,基于苹果Hsf蛋白全长序列构建了该家族的进化树(图2-A)。一般而言,位于进化树末端的同源基因对具有相同或相似的序列特征,例如MdHsfA2a/b、MdHsfA9c/d、MdHsfA4a/b、MdHsfB4a/b和MdHsfB2b/c等(图2-B);但也有许多同源基因对的序列特征发生了较大的分化,尤其是基因结构,其差异非常明显,例如MdHsfA6a/b、MdHsfA6c/e、MdHsfA10a/b、MdHsfA10e/f和MdHsfC1a/b等(图2-B)。与基因结构相比,同源基因对的保守基序和功能结构域的组成与排列顺序几乎完全相同(图2-C、D),具有非常高的保守性,这意味着内含子序列的变异是驱动基因分化的主要动力。
2.3 苹果Hsf基因的表达谱分析
在50个MdHsf家族成员中,MdHsfA2a、MdHsfA3a/b/c、MdHsfA6c/d/e、MdHsfA9a/b/e、MdHsfA10c/e/g和MdHsfC1a共有14个基因未找到与Hsf基因编码序列显著匹配的EST序列,因此,这些基因是否具有转录活性有待进一步试验验证。其余的36个基因均具有转录活性,占基因总数的72%。由于MdHsfA5a/b、MdHsfB3a/b和MdHsfA10b相应的UniGene中EST并没有进行正态化和统计处理,因此,不能推断这些基因的表达水平。根据这些基因相应的EST或cDNA的组织器官来源,可推知:MdHsfA5a/b在花和果实中均有转录活性,MdHsfB3a/b在花中有转录活性,MdHsfA10b在果实中有转录活性。由于有些苹果Hsf基因之间相似程度高,多个基因同时对应1个UniGene,因此,31个苹果Hsf基因只能检测到13个UniGene。这些UniGene的EST表达谱结果显示:在苹果根中,MdHsfB1a/b/c、MdHsfA4d/e和MdHsfC1a/c等基因均有表达,其中MdHsfA4d/e基因表达量最高;在苹果茎中,MdHsfA4a/b/c、MdHsfA8a/b/c、MdHsfA9c/d/g、MdHsfB1a/b/c和MdHsfB2b/c/d等基因均有表达,其中B亚家族基因MdHsfB1a/b/c和MdHsfB2b/c/d表达量最高;在苹果叶中,除了MdHsfA4a、MdHsfA9c/d、MdHsfB4a/b和MdHsfC1a/c等基因没有转录活性外,其他基因均有表达,其中MdHsfB1a/b/c表达量最高;在苹果花中,所有基因均没有转录活性;在苹果果实中,MdHsfA4d/e、MdHsfA9c/d/g、MdHsfB1a/b/c、MdHsfB2b/c/d、MdHsfA10f和MdHsfC1a/c等基因均有表达,其中MdHsfA4d/e表达水平最高;在苹果芽中,MdHsfA8a/b/c、MdHsfB1a/b/c和MdHsfB4a/b等基因均未检测到转录活性;在苹果细胞培养组织中,只有MdHsfB1a/b/c等基因具有转录活性(图 3)。

图2 基于苹果Hsf蛋白序列的进化树及Hsf基因及其蛋白的序列特征
2.4 基于位点模型的正选择位点检测
M0和M3、M1a和M2a、M7和M8是3对位点特异模型,这些模型假设了ω值在不同分支之间是同质的,在不同位点是异质的。因此,它们常被用于检测基因不同位点的选择压力。首先,准备序列比对文件和无根树(图4),然后用PAML软件包中的Codeml程序对36个苹果Hsf基因进行选择压力分析,进一步利用LRT测试所鉴定的正选择位点是否达到显著水平。结果显示:与相应的假设模型M1a和M7相比,备择模型M2a和M8均不具有优势,这一结论受到LRT检测的支持;尽管M3和M0之间的LRT检测支持苹果Hsf基因经历快速进化,但是这个模型对不推荐作为参考标准(表2)。这一结果揭示苹果Hsf基因在进化过程中受到了负选择,具有较高的保守性。
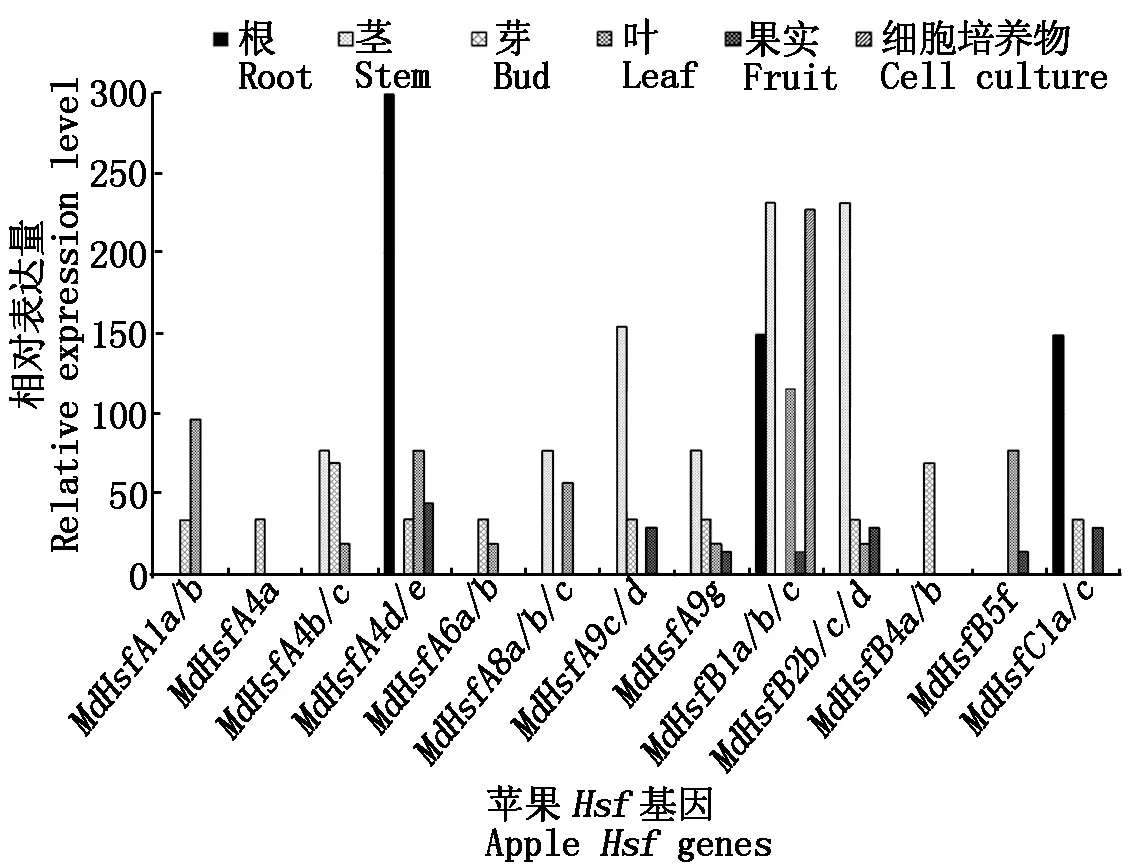
图3 MdHsf家族基因在苹果不同器官中的表达分析
2.5 基于分支-位点模型的正选择位点检测
在显著水平上,位点模型没有检测到正选择位点,这有可能是该模型并不适合于苹果Hsf基因。由于分支-位点模型允许不同分支上不同位点具有不同的功能约束和进化速率,这就意味着不同分支不同位点的ω值具有异质性,因此,该模型可以评价不同分支上不同位点所受到的选择压力。
将所有包括2个或2个以上基因的进化支标记为前景支,剩余的其他分支标记为背景支,执行检测时用Model A的测验2[29-30]。结果显示:以a、b、c、d、e、f、g进化支为前景支时,它们的ω值均大于1,除了c进化支没有检测到正选择位点以外,其余进化支均能检测到此类位点;尽管a、b、f、g进化支的ω值大于1,并且能够检测到正选择位点,但在显著水平(P<0.05)上,LRT检测并不支持这些正选择位点的存在;d、e进化支为前景支时,它们不但ω值大于1,而且在显著水平上,d、e进化支中分别包括3,2个正选择位点(图 4、表 3)。
2.6 苹果热激转录因子三维结构的建模
为阐明苹果Hsf蛋白的立体结构以及正选择位点在三维空间中的位置,以位于进化分支d和e中MdHsfA4e/d和MdHsfC1a/b蛋白作为研究对象,利用SWISS-MODEL工具对这4个蛋白结构进行同源模拟,经Loop区优化、能量最小化和动力学模拟分别得到最佳三维构象(图5)。结果显示:MdHsfA4d(图5-A)、MdHsfC1a(图5-C)和MdHsfC1b(图5-D)共享同一模板2lduA,它们与模板匹配序列区段的相似性分别为44.25%,42.48%,43.24%,它们的结构极其相似,主要结构包括3个α-螺旋、4个β-折叠和7或8个β-转角;MdHsfA4e(图5-B)的模板是3hsfA,其序列匹配序列区段的相似性为45.59%,它的结构明显有别于另外3个蛋白结构,包括3个α-螺旋、2个β-折叠和5个β-转角。由于模拟蛋白是部分序列的三维结构,而该序列C端包括正选择位点28R和30L,但不包括正选择位点35D、51M和67V,这说明该蛋白片段的C端在d和e进化支上均发生了快速进化。

粗线表示进化支的ω值大于1;箭头表示进化支的ω值大于1且具有统计意义上的显著性。
The thick line represents the ω value of the evolution branch is greater than 1;The arrow indicates that the ω value of the evolution branch is greater than 1 and also is significant statistically.
图4 用于苹果Hsf蛋白选择压力检测的无根树
Fig.4 The unrooted tree used in detection for selection pressures on Hsf proteins in apple

表2 基于位点模型的苹果Hsf基因正选择位点检测
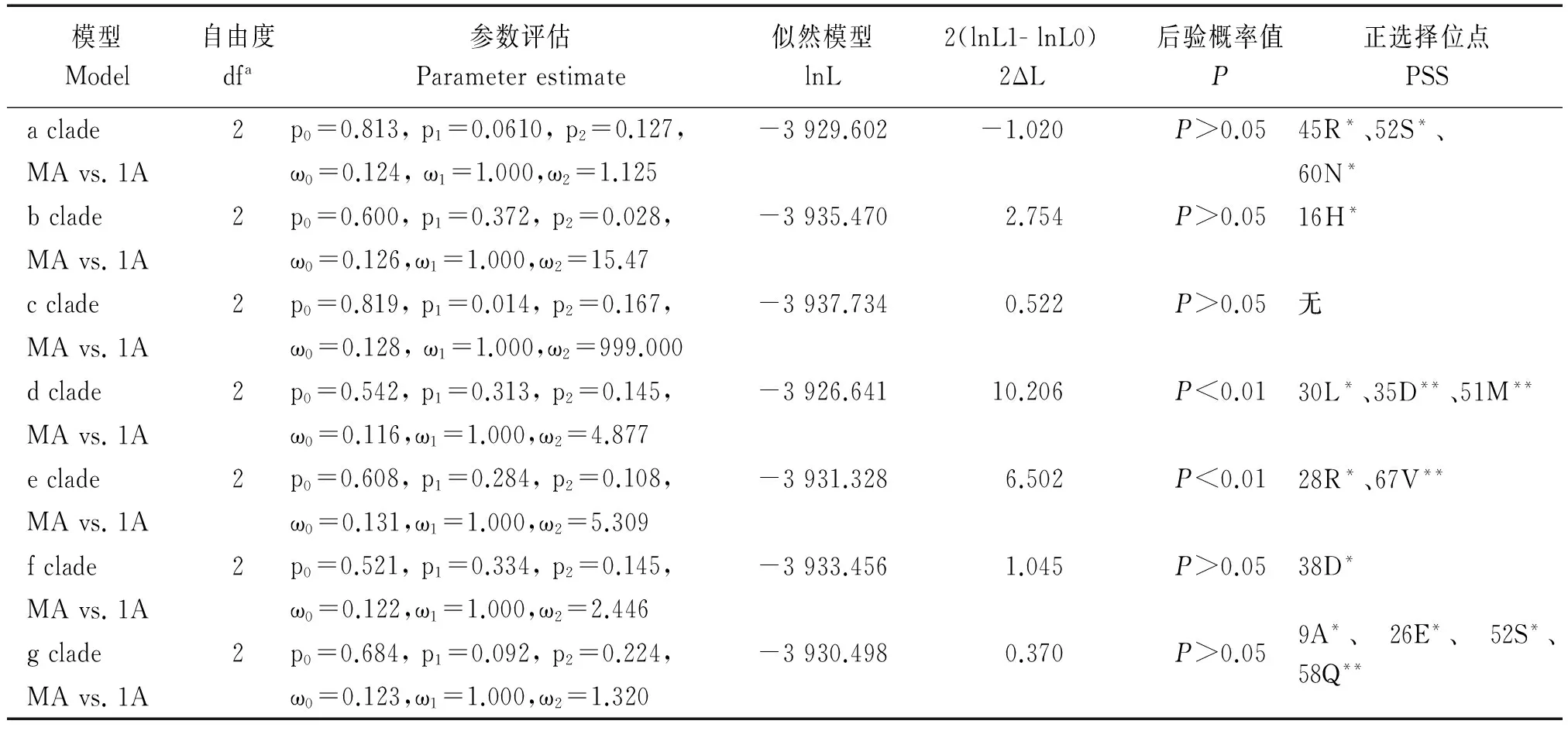
表3 基于分支-位点模型的苹果Hsf基因选择压力检测
3 结论与讨论
基于生物信息学手段,本研究以Hsf保守结构域为检索序列,鉴定了苹果基因组中具有50个Hsf基因家族的成员,这比Giorno等[20]鉴定的成员数目多了1倍,这可能是由于所采用的鉴定方法有所不同。从基因结构、蛋白结构和进化分析上来看,笔者鉴定的基因都为Hsf基因家族的成员,所以,本研究是在苹果全基因组水平上全面而系统的对Hsf基因家族的分析。根据系统分析结果,并参考在其他物种中Hsf基因的分类情况,将苹果Hsf基因家族分为A、B和C 3个亚家族。该分类结果与Giorno等[20]的分类是一致的。目前在所研究的物种中,对于HSF的分类一般都采用A、B、C 3个亚家族的分类,其中B一般会形成单系,C在一些物种中会形成单系,在另外的物种是和A亚家族聚在一起的,而A亚家族包含的成员数量众多,一般很少会形成单系[17,31-32]。系统进化分析显示,2个或多个苹果Hsf蛋白总是先聚在一起,然后与一个拟南芥同源基因再聚在一起,最后与水稻相应同源基因聚为一簇,这不仅说明Hsf基因在苹果基因组中发生了扩增,也印证了这3个物种的演化过程。苹果基因组测序分析揭示了该物种是由其祖先物种(与灭绝物种Gillenia相似)全基因加倍后,再经过二倍和非整倍体化过程,然后形成的新物种[19],因此,苹果Hsf基因的扩增与全基因组加倍具有十分密切的关系。由于热激转录因子特异识别热激蛋白启动子区的保守顺式元件(HSE:AGAAnnTTCT),因此,它至少包括能与HSE元件特异结合的功能结构域。通常情况下,植物热激转录因子主要包括:1个N端DNA结合区域(DNA binding domain,DBD)、1个双向寡聚化区域(Heptads repeat of hydrophobic amino acid residues,HR-A/B)、1个细胞核定位信号(Nuclear localization signal,NLS)和细胞核输出信号(Nuclear export signal,NES)[16];此外,少数植物Hsf蛋白还具有1个酸性C端的激活域(C terminal activator domain,CTAD)[16]。从基因结构角度看,苹果Hsf基因具有多样化的结构,尤其是内含子数目和长度,存在着丰富的变异。从蛋白水平看,该蛋白家族成员之间具有较高的相似性,这是因为Hsf蛋白保守基序与功能结构域之间相互重叠,例如DBD与Motif1/2,HR-A/B与Motif3/4等。Hsf蛋白为了正确行使功能,其序列的变异就会受限制,因而蛋白序列具有较高的保守性,但不同亚家族之间保守基序组成类型可能不同。

图5 四个苹果热激转录因子蛋白的结构
就苹果Hsf基因家族成员的表达而言,虽然UniGene中的EST或cDNA序列能推断基因的转录活性,甚至表达的相对量,但是基于同一个UniGene的EST表达谱推断多个同源基因表达是不精确的。这是因为进化过程中,作为非编码区的启动子发生变异较快,而启动子序列又是基因表达调控的关键元件,因此,同源基因的表达或多或少会有差异,其精确表达有待试验进一步验证。
在选择压力检测时,通常用ω=dN/dS值来衡量选择压力。若ω>1且似然比检验具有显著性差异,则认为编码序列在对应的分支或位点经受正选择。本研究采用位点模型与分支-位点模型,针对36个苹果Hsf基因进行选择压力分析。尽管位点模型没有检测到正选择位点的存在,但分支-位点模型在d和e进化支上共检测到5个正选择位点,其中d进化支上正选择位点是30L、35D和51M,e进化支上是28R和67V。为了确定这5个正选择位点在Hsf蛋白三维结构中的位置,本研究在模拟了MdHsfA4d、MdHsfA4e、MdHsfC1a和MdHsfC1b(图5-D)蛋白结构的基础上,比较了正选择位点与模拟结构区序列。结果显示,d进化支上的30L和e进化支上的28R均被定位于保守的Hsf结构域中,这说明Hsf结构域不仅高度保守,而且在特定进化支的某些位点也发生了快速进化。D进化支上的35D和51M以及e进化支上的67V正选择位点均远离活性DNA结合区域(DNA binding domain,DBD),并且它们分布在不同的位置,这充分说明了正选择很可能提高该蛋白家族特定进化支的适应性。总之,纯净选择主导了该家族的进化,尤其对于Hsf结构域而言,纯净选择是该蛋白行使功能,维持酶活性的基础。同时揭示快速进化可以发生特定进化支系的某些位点内,很可能为该酶结构演化和适应新环境提供原始动力。本研究结果为后续的Hsf功能研究和利用基因工程方法改良蛋白活性提供参考信息。
[1] Åkerfelt M, Morimoto R I, Sistonen L. Heat shock factors: integrators of cell stress, development and lifespan[J]. Nat Rev Mol Cell Bio, 2010, 11(8): 545-555.
[2] Ohama N, Sato H, Shinozaki K, et al. Transcriptional regulatory network of plant heat stress response[J]. Trends Plant Sci, 2016, 22(1): 53-65.
[3] Wiederrecht G,Seto D,Parker C S.Isolation of the gene encoding theS.cerevisiaeheatshock transcription factor [J]. Cell,1988,54(6):841-853.
[4] Clos J,Westwood J T,Becker P B,et al.Molecular cloning and expression of a heaxameric drosophila heat stress factor subject to negative regulation [J].Cell,1990,63(5):1085-1097.
[5] Fujimoto M, Hayashida N, Katoh, et al. A novel mouse HSF3 has a potential to activate nonclassical heat-shock genes during heat shock[J]. Mol Biol Cell, 2010, 21(1): 106-116.
[6] Rabindran S K,Giorgi G,Clos J,et al.Molecular cloning and expression of a human heat shock factor,HSF1 [J].Proc Natl Acad Sci USA,1991,88(16):6906-6910.
[7] Scharf K D,Rose S,Zott W,et al.Three tomato genes code for heat stress transcription factors with a region of remarkable homology to the DNA-binding domain of the yeast HSF [J].EMBO J,1990,9(13):4495-4501.
[8] Hü bel A,Schö ffl F.Arabidopsisheat shock factor:isolation and characterization of the gene and the recombinant protein [J].Plant Mol Biol,1994,26(1):353-362.
[9] Yamanouchi U,Yano M,Lin H,et al.A rice spotted leaf gene,Spl7,encodes a heat stress transcription factor protein [J].Proc Natl Acad Sci USA,2002,99(11):7530-7535.
[10] Guo J,Wu J,Ji Q,et al.Genome-wide analysis of heat shock transcription factor families in rice andArabidopsis[J].J Genetics Genomics,2008,35(2):105-118.
[11] Chauhan H,Khurana N,Agarwal P,et al.Heat shock factors in rice(OryzasativaL.):genome-wide expression analysis during reproductive development and abiotic stress [J].Mol Genet Genomics,2011,286(2):171-187.
[12] Lin Y X,Jiang H Y,Chu Z X,et al.Genome-wide identification,classification and analysis of heat shock transcription factor family in maize [J].BMC Genomics,2011,12(1):76.
[13] Scharf K D,Berberich T,Ebersberger I,et al.The plant heat stress transcription factor(Hsf)family:structure,function and evolution [J].Biochimica et Biophysica Acta 2012,1819(2):104-119.
[14] Song X,Liu G,Duan W,et al.Genome-wide identification,classification and expression analysis of the heat shock transcription factor family in Chinese cabbage [J].Mol Genet Genomics,2014,289(4):541-551.
[15] Huang Y,Li M Y,Wang F,et al.Heat shock factors in carrot:genome-wide identification,classification,and expression profiles response to abiotic stress [J].Mol Biol Rep,2015,42(5):893-905.
[16] Liu Z W, Wu Z J, Li X H, et al. Identification, classification, and expression profiles of heat shock transcription factors in tea plant (Camelliasinensis) under temperature stress[J]. Gene, 2016, 576(1): 52-59.
[17] Guo M, Liu J H, Ma X, et al. The plant heat stress transcription factors (HSFs): structure, regulation, and function in response to abiotic stresses[J]. Front Plant Sci, 2016, 7(273): 114.
[18] Raxwal V. Structural and functional diversity of plant heat shock factors[J]. Plant Stress, 2012, 6: 89-96.
[19] Velasco R,Zharkikh A,Affourtit J,et al.The genome of the domesticated apple(MalusdomesticaBorkh.)[J].Nature Genetics,2010,42(10):833-839.
[20] Giorno F,Guerriero G,Baric S,et al.Heat shock transcriptional factors inMalusdomestica:identification,classification and expression analysis [J].BMC Genomics,2012,13(1):639.
[21] Edgar R C.MUSCLE:multiple sequence alignment with high accuracy and high throughput [J].Nucl Acids Res,2004,32(5):1792-1797.
[22] Tamura K, Stecher G, Peterson D, et al. MEGA6: Molecular evolutionary genetics analysis version 6.0[J]. Mol Biol Evol, 2013, 30(12): 2725-2729.
[23] Thompson J D,Gibson T J,Plewniak F,et al.The CLUSTAL_X windows interface:flexible strategies for multiple sequence alignment aided by quality analysis tools [J].Nucleic Acids Res,1997,25(25):4876-4882.
[24] Castresana J.Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis [J].Mol Biol Evol,2000,17(4):540-552.
[25] Suyama M,Torrents D,Bork P.PAL2NAL:robust conversion of protein sequence alignments into the corresponding codon alignments [J].Nucleic Acids Res,2006,34:609-612.
[26] Page R D.TreeView:an application to display phylogenetic trees on personal computers [J].Comput Appl Biosci,1996,12(4):357-358.
[27] Yang Z.PAML4:phylogenetic analysis by maximum likelihood [J].Mol Biol Evol,2007,24(8):1586-1591.
[28] Arnold K,Bordoli L,Kopp J,et al.The SWISS-MODEL workspace:a web-based environment for protein structure homology modelling [J].Bioinformatics,2006,22(2):195-201.
[29] Yang Z,Wong W S,Nielsen R.Bayes empirical Bayes inference of amino acid sites under positive selection [J].Mol Biol Evol,2005,22(4):1107-1118.
[30] Zhang J,Nielsen R,Yang Z.Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level [J].Mol Biol Evol,2005,22(12):2472-2479.
[31] Wang F,Dong Q,Jiang H,et al.Genome-wide analysis of the heat shock transcription factors inPopulustrichocarpaandMedicagotruncatula[J].Mol Biol Rep,2012,39(2):1877-1886.
[32] Chung E,Kim K M,Lee J H.Genome-wide analysis and molecular characterization of heat shock transcription factor family inGlycinemax[J].J Genet Genomics,2013,40(3):127-135.
Sequence Characterization,Expression,and Evolutionary Analysis of Heat Shock Transcription Factors in Apple
ZHANG Guojun1,2,WANG Tingting3,HU Lizong2,LI Shufen2,GAO Wujun2
(1.Scoool of Basic Medical Sciences,Xinxiang Medical University,Xinxiang 453003,China;2.College of Life Sciences,Henan Normal University,Xinxiang 453007,China;3.Department of Food Engineering,Luohe Vocational Technology College,Luohe 462000,China)
To extensively understand the sequence feature and evolution of heat shock transcription factors(Hsf)in the genome of apple,fiftyMdHsfgenes were identified using bioinformatics methods at the whole-genome level of apple,and a series of analysis including sequence characterization,phylogenetic relationship,gene expression and selective pressure ofMdHsfgenes were further performed.Phylogenetic relationship and sequence characterization analysis showed that,like the model speciesArabidopsisand rice,50MdHsfgenes were divided into three subfamilies A,B and C.Additionally,at least two genes were found in the same end clades in the phylogenetic tree,indicating that the lineage-specific amplification had happened during evolutionary processes of appleHsfgene family.Although the intron numbers and sizes ofMdHsfgenes were relatively divergent,the conserved motifs and domains of MdHsf proteins were highly conserved because of functional constraints.Based on EST data,72% of the 50 genes(except 14 genes such asMdHsfA2aandMdHsfA3a/b/c)had transcription activities.Selective pressure signatures demonstrated that no positive selection site was identified in the cleaned codon alignments for 36MdHsfgenes based on site-specific model,suggesting that this protein family was controlled by purifying selection.However,branch-site model had identified a total of five positively selected sites in the d and e clade of the phylogenetic tree,i.e.28R,30L,35D,51M and 67V.28R and 30L were included in the Hsf domains,while 35D,51M and 67V were not mapped on the region of Hsf domains,suggesting that purifying selection was the main evolutionary dynamics of functional conservation Hsf domains except for 28R and 30L.In conclusion,various Hsfs existed in apple genome,and the conserved motifs and functional domains were conserved.The majority of them had transcription activity,and the evolution of this family was dominated by purifying selection.
Apple;Heat shock transcription factors(Hsf);Expression;Evolution
2016-08-12
国家自然科学基金项目(31300202;31470334)
张国俊(1980-),男,河南南阳人,讲师,硕士,主要从事分子遗传学研究。
高武军(1973-),男,山西芮城人,教授,博士,主要从事分子细胞遗传学研究。
Q78;S661.03
A
1000-7091(2017)02-0071-10
10.7668/hbnxb.2017.02.012
猜你喜欢
湖北农业科学(2022年11期)2022-07-18
南方医科大学学报(2022年3期)2022-04-13
浙江大学学报(农业与生命科学版)(2021年3期)2021-07-10
内蒙古师范大学学报(自然科学汉文版)(2021年3期)2021-06-01
实用肿瘤学杂志(2020年4期)2020-12-08
中国预防兽医学报(2020年8期)2020-11-05
三农资讯半月报(2020年15期)2020-08-25
医药前沿(2019年18期)2019-01-04
中国棉花(2018年6期)2018-07-06
中国老年学杂志(2015年9期)2015-01-31
