
不同氮处理下水稻剑叶叶宽的全基因组关联分析
2017-07-31 23:47高易宏燕金香涂政军冷语佳陈龙黄李超代丽萍张光恒朱丽胡江任德勇郭龙彪钱前王丹英曾大力
中国农业科学 2017年14期
高易宏,燕金香,涂政军,冷语佳,陈龙,黄李超,代丽萍,张光恒,朱丽,胡江,任德勇,郭龙彪,钱前,王丹英,曾大力
不同氮处理下水稻剑叶叶宽的全基因组关联分析
高易宏,燕金香,涂政军,冷语佳,陈龙,黄李超,代丽萍,张光恒,朱丽,胡江,任德勇,郭龙彪,钱前,王丹英,曾大力
(中国水稻研究所/水稻生物学国家重点实验室,杭州 310006)
【目的】探索水稻剑叶叶宽调控及其对氮肥响应的遗传机理,为氮高效水稻新品种的培育提供新的种质资源和基因标记。【方法】以134份水稻地方种质资源为关联分析材料,通过基因组重测序发掘获得了3 356 591个分布于全基因组的高密度SNP位点(single nucleotide polymorphism,SNP)。在大田栽培条件下,以施氮量为主区,品种为裂区的设计,设计低氮(不施氮肥,N0)、正常氮(纯氮96 kg·hm-2,N1)和高氮(纯氮192 kg·hm-2,N2)3种氮肥处理。于水稻成熟期分别调查水稻剑叶叶宽在低、中、高3种氮肥处理下的表现及响应,结合EMMAX软件计算群体亲缘关系矩阵和EIGENSOFT软件分析群体结构,采用纳入亲缘关系矩阵及群体结构的混合线性模型开展全基因组关联分析。【结果】水稻剑叶叶宽在N0、N1、N2 3种氮肥处理下均呈正态分布,并表现丰富的变异。剑叶叶宽受品种差异及氮水平的影响,且与施氮水平呈显著正相关。在N0、N1、N2氮处理下共检测到14个与剑叶叶宽显著相关的SNP位点。其中,低氮处理下检测到的SNP位点的最小等位基因频率均大于0.46,表明此类SNP在关联群体中广泛存在;中氮和高氮水平下检测到的SNP位点的最小等位基因频率均较小,是一类较为稀有的SNP位点。位于第12染色体上的一个SNP(chr12:15 066 507)位点在正常氮及高氮处理下均被检测到,在高氮处理下还检测到的另一显著性位点,其候选区间内包含一候选基因,该基因与业已报道的叶宽性状相关基因同属于细胞色素P450家族。根据不同氮处理下剑叶叶宽的响应,鉴定出20与低氮响应有关的SNP位点,8个位点与高氮响应有关。其中与高氮响应的显著性位点中,位于第1染色体显著性峰候选区间包含业已克隆的与氮素利用相关的基因。【结论】通过全基因组关联分析共检测到42个与剑叶叶宽及其在不同氮处理下叶宽响应相关的显著性关联位点。
水稻;全基因组关联分析;叶宽;氮肥
0 引言
【研究意义】氮是植物生长发育中至关重要的营养元素,合理的氮肥施用将显著的增加作物产量[1],氮营养供应不充足则会导致作物减产,而过量施用氮肥会导致环境污染等问题。因而氮素的高效利用是当前水稻育种关注的重要问题,通过选育氮高效吸收利用的水稻品种,从而减少田间氮肥施用以提高水稻产量已成为水稻育种的重要目标之一[2]。【前人研究进展】水稻的氮素利用效率(nitrogen use efficiency,NUE)受品种、氮肥用量、氮肥施用方法和土壤含氮量等因素的影响。近年来,通过对水稻氮响应相关突变体的研究和QTL定位等方式克隆与鉴定了多个与NUE相关的基因及位点。Zhang等[3]鉴定了一个位于第12染色体上有关氮耐受性的QTL位点,并克隆了。Hu等[4]通过在高世代回交群体分离鉴定了一个硝酸盐转运基因,该基因通过增强硝酸盐的摄取、根向茎转运和氮响应基因的表达从而提高氮素利用率。Sun等[5]通过分析RIL群体在不同氮浓度下株高和分蘖数的变化鉴定了一个水稻主效的NUE数量性状位点,实为已报道的直立密穗基因,携带的植株表现出对氮吸收和同化的增强,从而增加水稻产量。在长期的进化过程中,植物形成了适应不同生存环境的生理机制和分子机理来响应环境的变化。在应对缺氮环境时,植物提高NUE的主要方式是通过提高氮的吸收、转运、同化和再活化等方式,且与环境之间存在复杂的互作及遗传网络关系[6-9]。Tong等[10]通过在2种氮处理条件下的株高、穗数、叶绿素含量、干重、湿重和产量变化等指标鉴定了31个氮响应相关的QTL位点。Cho等[11]在两种氮浓度下,通过水稻发育后期籽粒中氮含量、秸秆中氮含量、收获指数等多个指标鉴定了多个氮响应相关的QTL位点。同样,在不同的氮水平下水稻叶片呈现明显的叶形变化,而水稻叶片的长、宽和厚的改变也直接影响水稻的光合作用[12]。殷春渊等[13]研究表明,氮肥处理对水稻叶片的影响主要表现在倒二叶叶长和剑叶叶宽2个特性上。因此,叶片形态结构的变化也是水稻对不同水平氮响应的指标。近年来,全基因组关联分析(genome-wide association study,GWAS)成功的应用在一些模式生物研究和农作物的育种当中[14~18]。【本研究切入点】在水稻中,利用GWAS分析了包括产量、品质、抽穗期、株形等多种重要的农艺性状,同样也对病虫害抗性等生物胁迫和盐碱耐受性等非生物胁迫性状进行了全基因组关联分析,并鉴定了许多重要的SNP位点[14,16-17,19-22]。但是,与氮素利用相关的GWAS研究尚未见报道。【拟解决的关键问题】本研究通过选取134份来自不同地区的水稻品种作为关联分析的群体,利用在不同氮肥条件下叶宽的变化,开展水稻叶宽及氮响应相关SNP位点分析,旨在为进一步揭示水稻叶片形态调控和氮素高效利用的生理机制提供基础。
1 材料与方法
1.1 试验材料
选择保存于中国水稻所(CNRRI),国际水稻所(IRRI)种质资源库及来自多个国家和地区的134个地方品种,其中包括41份来自国内各省农科院的地方品种;36份来自20个国家的地方品种;57份国内大面积推广种植品种。
1.2 试验设置及性状考察
试验于2015年在中国水稻研究所实验基地(浙江,富阳)进行,试验田土壤为水稻土(土壤含有机质36.9 g·kg-1、全氮2.73 g·kg-1、速效钾104 mg·kg-1、速效磷52 mg·kg-1、碱解氮156 mg·kg-1,pH 6.24)。试验设3个氮肥水平:低氮(N0,不另施氮肥)、正常氮(N1,纯氮96 kg·hm-2)和高氮(N2,纯氮192 kg·hm-2),以施氮量为主区,品种为裂区,3次重复。主区面积5.0 m×23.0 m,播种前将其做成3条畦宽1.5 m,沟宽25 cm,长23 m的畦。水稻种于畦上,每品种种植4行,每行6株,株行距为20 cm×20 cm。不同氮处理间作埂隔离,并用塑料薄膜覆盖埂体,保证单独排灌。
试验用关联群体于2015年5月10日采用点直播栽培,每穴播3—5粒种子,2叶1心时定苗2株/穴。以尿素为氮肥,按基肥﹕分蘖肥﹕穗肥 = 5﹕3﹕2分3次施入,其中基肥(播种前1 d)与分蘖肥(播种后25 d)统一施用,穗肥由于品种间生育期的差异,于孕穗始期分品种单独施用。6月中旬至9月中旬每隔3d检查品种的生育期,标记孕穗始期相近的品种,将穗肥小心撒于每个标记品种叶片上。为防止水分流动导致的串肥,穗肥施用期间(6月中旬至9月中旬)保持畦面湿润但无水层,仅沟中有水。磷、钾施用量为P2O575 kg·hm-2、K2O 150 kg·hm-2,其中磷肥为底肥一次施入,钙、镁、钾肥按基、蘖肥各50%分2次施入。其他栽培管理措施同一般大田。
在抽穗后10 d对剑叶叶宽进行调查,每品种选取中间的6株无边际效应的植株进行考察。参考王兰等[23]方法量取每片叶的最宽处为叶宽,每株取主茎分别记录剑叶的叶宽。
1.3 数据分析
1.3.1 性状相关性分析 使用Excel 2010进行数据的录入和整理,并计算其平均值和标准差。使用SAS 9.2进行进行不同氮处理下叶宽变化的相关分析和方差分析。
1.3.2 基因型测定 采用CTAB抽提法提取134份品种幼叶基因组DNA[24],检测合格后用于高通量测序。每品种测序reads数据量在5Gb以上,相当于10×以上的基因组测序。从reads到SNP的检测参考Zhou等[17]方法进行。SNP使用ANNOVAR (Version: 2013-08-23)软件包进行注释[25]。
1.3.3 群体结构与亲缘关系分析 根据SNP信息,使用PHYLIP 3.68软件包基于邻接法构建品种间系统进化树[26],使用在线工具iTOL(http://itol.embl.de/)进行进化树的优化。使用EIGENSOFT软件中的smartpca程序包进行主成分分析(PCA)[27]。
1.3.4 叶宽性状的全基因组关联分析 采用基于混合线性模型(mixed linear model,MLM)的EMMAX(Efficient Mixed-Model Association eXpedited)软件包进行目标性状与SNP标记间的关联分析[20, 28],将EMMAX软件计算的亲缘关系K矩阵及基于EIGENSOFT软件计算的群体结构Q纳入分析模型中,关联分析参数设定为:emmax -v,-d 10,-t,-p,-k,-c,-o。在考虑关联分析群体大小和SNP数量的情况下,对于关联分析的显著性,采取10-6作为宽松阈值,而进一步的严格阈值采取Bonferroni校验阈值[29]。
2 结果
2.1 3种氮处理条件下叶宽性状多样性及其变化
在134份材料中,剑叶叶宽表现丰富的变异,正常氮处理下变幅为9.93—29.00 mm。如木邦谷、罗大穗等地方品种表现为株型较大,分蘖少,成熟期叶片少,剑叶叶宽大于20 mm;而地方品种英德大叶拍、合江23等表现为株型小,成熟期叶片多,叶片呈细长状,叶宽小于10 mm。由此表明,此关联群体具有充分的表型变异,能够进行基于叶宽的全基因组关联分析。
对裂区设计整体进行方差分析,变异来源于氮肥使用量、品种及品种与氮肥交互作用的值均小于0.0001,而区组与氮处理间的交互作用为0.2444,这说明叶宽变异主要受氮处理及品种差异引起,不同品种在不同氮浓度下展现不同的叶宽变化。对134份品种成熟期剑叶宽调查结果表明,在低、中、高3种氮处理下,剑叶叶宽均呈正态分布(图1-a—图1-c),从频率分布来看,中氮和高氮处理下剑叶宽为15—18 mm的品种明显增多。在反映氮响应变化中,正常氮处理相对于低氮处理表现为叶宽变大,而相对于高氮处理变化要小(图1-d)。相关性分析亦表明,氮处理与剑叶叶宽呈显著正相关(表1),这表明,叶宽受外界氮水平的影响,且叶宽的变化能够反映不同氮处理。
根据不同氮处理下叶宽相对变化,将叶宽变化分为四种响应模式(图2-e)。有35.2%的品种表现为在高氮条件下叶宽增加而低氮条件下叶宽减小,有18.1%的品种在低氮和高氮条件下剑叶宽均增大,另有36.2%的品种在低氮和高氮条件下叶宽均减小,还有5.7%的品种在低氮条件下剑叶变宽而在高氮条件下反而变窄。由此可知,该关联群体在不同氮处理下叶宽相对变化呈现丰富的变异,可以利用叶宽的相对变化来反映氮素利用效率。
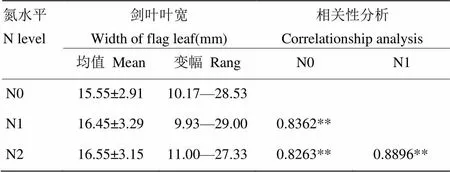
表1 不同氮浓度下叶宽统计及其相关性分析
*、**分别表示差异达到0.05 和 0.01 的显著水平
*,** present significant at 5% and 1% probability levels respectively
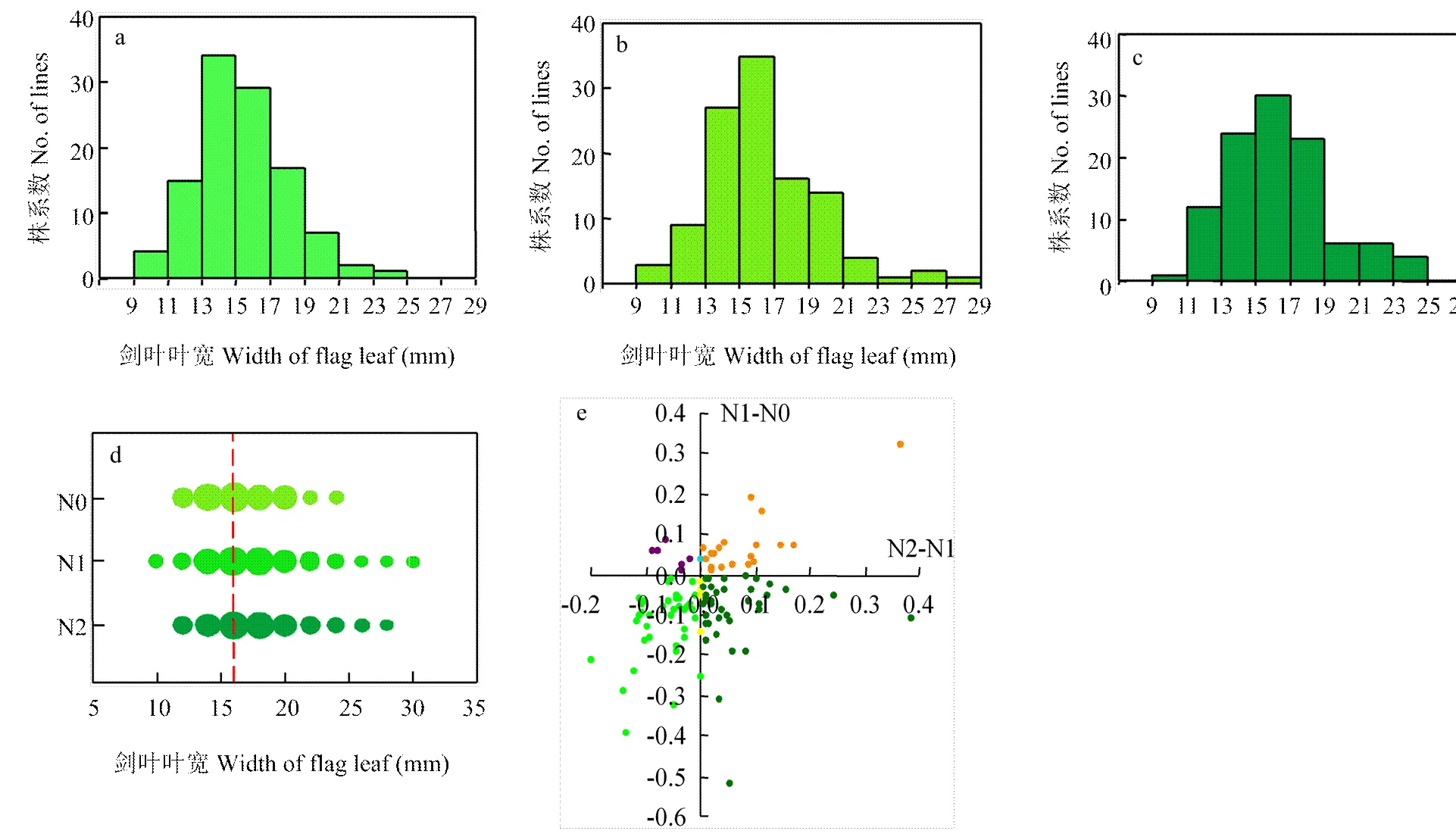
a、b、c分别为低氮、中氮和高氮条件下的叶宽分布;d:3种氮水平下叶宽分布的气泡图,气泡宽度代表株系数,红色虚线为3种氮条件下叶宽平均值;e:高氮胁迫下叶宽相对变化(N2-N1)和低氮胁迫下叶宽相对变化(N0-N1)的分布
2.2 关联分析SNP标记
基于134份水稻的测序数据,结合使用BWA和GATK软件进行SNP的提取,使用最小等位基因频率大于0.03(MAF>0.03)进行SNP的过滤,最终提取到3 356 591个SNP位点,平均每kb有8.6个SNP位点。为进一步分析SNP位点信息,使用ANNOVAR软件对SNP数据注释(图2),其中,有2 814个位点注释为剪接位点相关,此类位点通过影响基因转录剪接的变化导致基因功能改变;409 387个SNP位点注释到外显子区,引起核酸编码信息的改变,其中59%的SNP为非同义突变,造成编码信息的改变,从而引起蛋白序列的变化;另外有12 221个位点注释与终止密码子相关,终止密码子的缺失或增加会导致编码蛋白移码突变或提前终止。

图2 关联分析SNP标记分布及其功能注释
2.3 群体的遗传结构分析
基于134品种重测序的SNP数据,开展亲缘关系和系统进化分析。结果表明,此关联群体主要分为籼稻和粳稻两个亚群。由GAPIT软件基于SNP计算得到的亲缘关系系数表明,此134份水稻品种主要分为71份籼稻品种和63份粳稻品种,亚种内部分品种的亲缘关系较近,形成小的亚群(图3-a和图3-b)。基于SNP数据计算品种间遗传距离和NJ(Neighbor- joining)法构建系统发育树表明,此关联群体主要为籼粳之间的分歧(图3-c)。主成分分析(principal component analysis,PCA)同样也说明了此关联群体籼粳分化的群体结构。此外,基于PCA三点图可知,籼稻亚种间分化较为集中,粳稻亚种间存在不同程度的分化,较为分散,部分品种间存在小亚群结构(图3-d)。
2.4 叶宽性状的全基因组关联分析
通过对不同氮处理条件下134份水稻成熟期剑叶叶宽的全基因组关联分析表明,在不同氮处理条件下,与叶宽相关联的SNP位点各不相同(表2)。以-log10(p)大于6为阈值,共检测到14个与叶宽性状显著相关的SNP位点(图4)。在低氮和中氮水平下,均检测到6个显著的SNP位点与叶宽性状相关。而高氮水平只检测到位于第12染色体上的2个显著位点,其中一个位点与中氮水平一致,推测该调控叶宽的位点受氮水平影响较小。
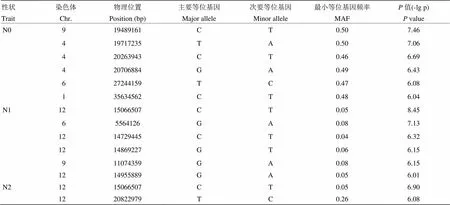
表2 3种氮处理下剑叶宽的关联位点
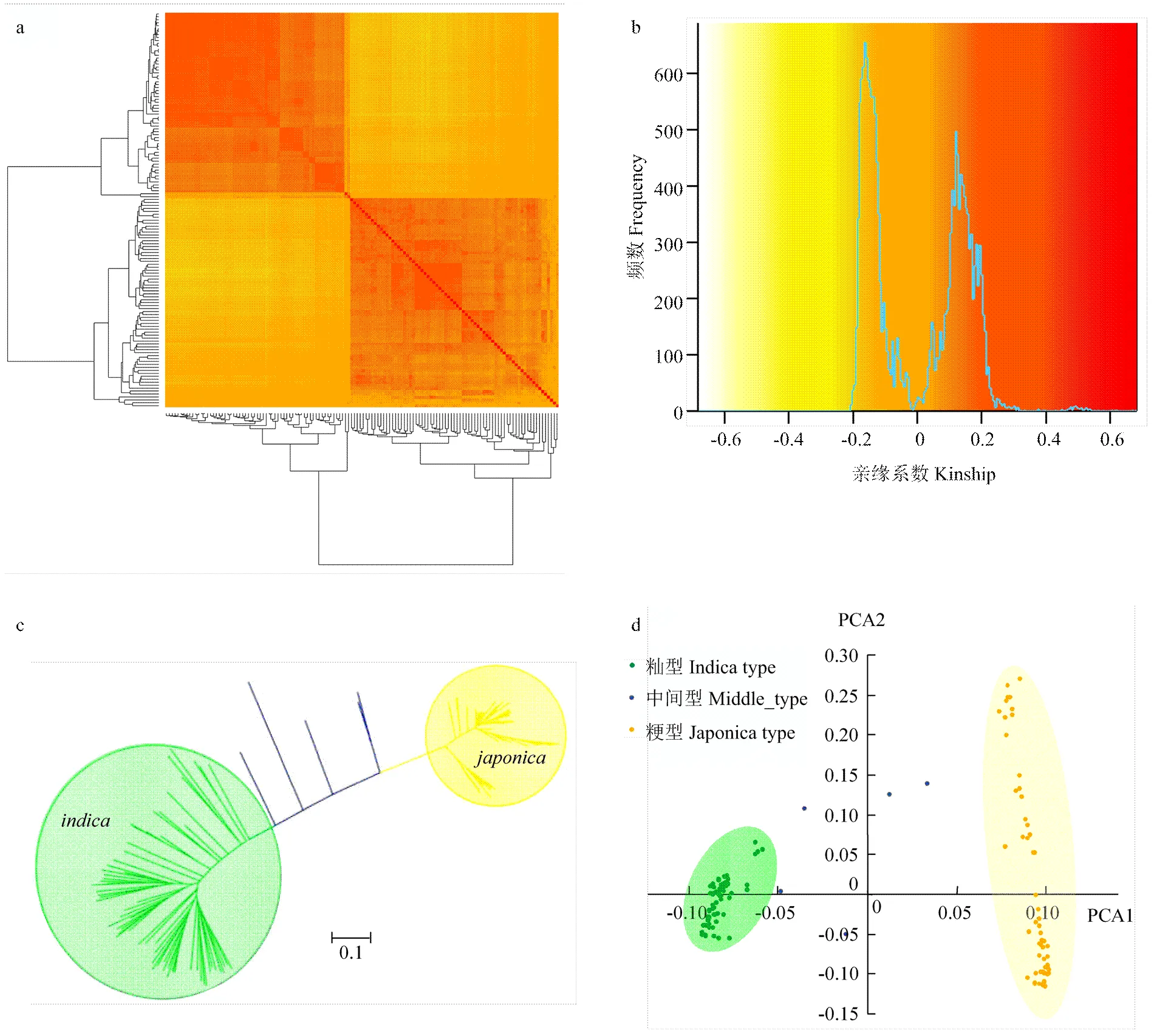
a、b:群体亲缘关系分布图;c:基于SNP标记构建的N-J树;d:主成分分析(PCA)散点分布图
在低氮水平下检测到的显著性SNP位点最小等位基因频率均大于0.46,此类SNP在关联群体中分布较为广泛。而在中氮和高氮水平下检测到的SNP位点最小等位基因频率均较小,属于一类较为稀有的突变位点。在中氮水平下,第12染色体上显著性SNP位点的值(-lgp)为8.45,高于Bonferroni校正后的阈值水平。对中氮水平下叶宽性状显著关联的位点进行候选基因分析时发现,位于第12染色体上的Chr12_14869227显著位点±200 kb的候选区间内,包含一注释为Cytochrome P450功能相关蛋白的候选基因,其与业已报道的叶宽性状相关基因同属于细胞色素P450家族(图5)。编码水稻BR生物合成途径中的一个关键的BR-6氧化酶,该酶活下降的植株表现为BR缺乏症状,而使叶宽受到显著的影响[30-31]。在高氮水平下,检测到另一个位于第12染色体上显著性关联的峰,该区段包含已知的,编码一个细胞壁生物合成和植株生长必须的糖基转移酶家族的纤维素合酶类似蛋白(cellulose synthase-like,CSL),的突变导致水稻叶片变窄、半卷和矮化等表型[32-33]。
2.5 不同氮水平剑叶叶宽的全基因组关联分析
进一步使用不同氮水平下叶宽的相对变化值来分析叶宽对氮水平的响应,共检测到28个显著的SNP位点,其中20个与低氮响应相关,8个与高氮响应相关(表3、图6)。在低氮环境下,叶宽的响应位点比高氮环境下多,这与叶宽变化在低氮环境下表型变异丰富,而在高氮水平下叶宽变化不明显有关。在高氮水平下,位于第7染色体上的显著性位点Chr7_20639350,其-lg(p)为9.01,超过了Bonferroni检验阈值;另外,还检测到位于第1染色体上的显著性峰,该区段存在多个显著性峰值点(图7),在此区段内存在一个已知的,该基因与氮素利用相关,表现为叶片明显早衰,氮的再活化作用被抑制[34-35],推测该基因为此检测区间的主效基因。

左侧为曼哈顿分布点图,右侧为Q-Q图;红色虚线显示为N1和N2水平下位于第12染色体共同的关联位点。下同
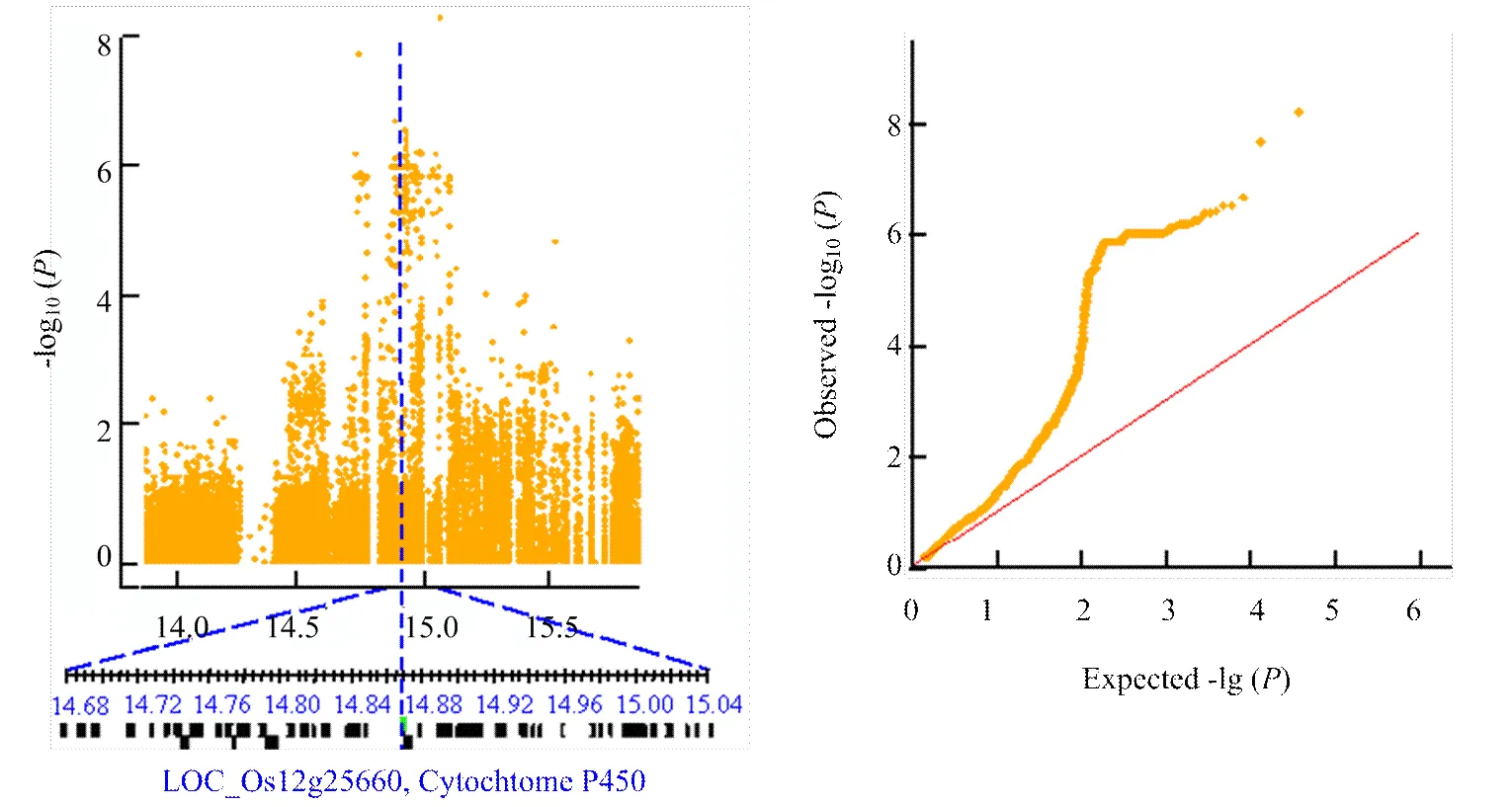
图5 第12染色体上在N1水平下的关联显著位点候选区间分析
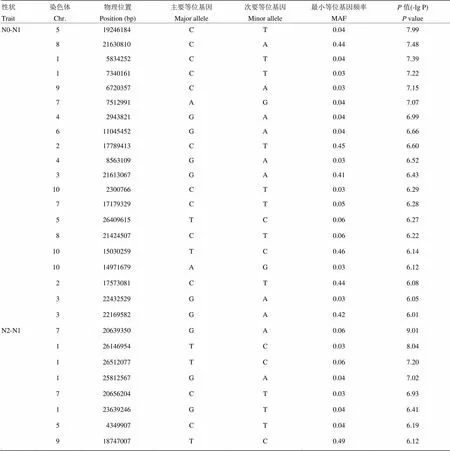
表3 低氮和高氮水平下剑叶叶宽变化的关联位点
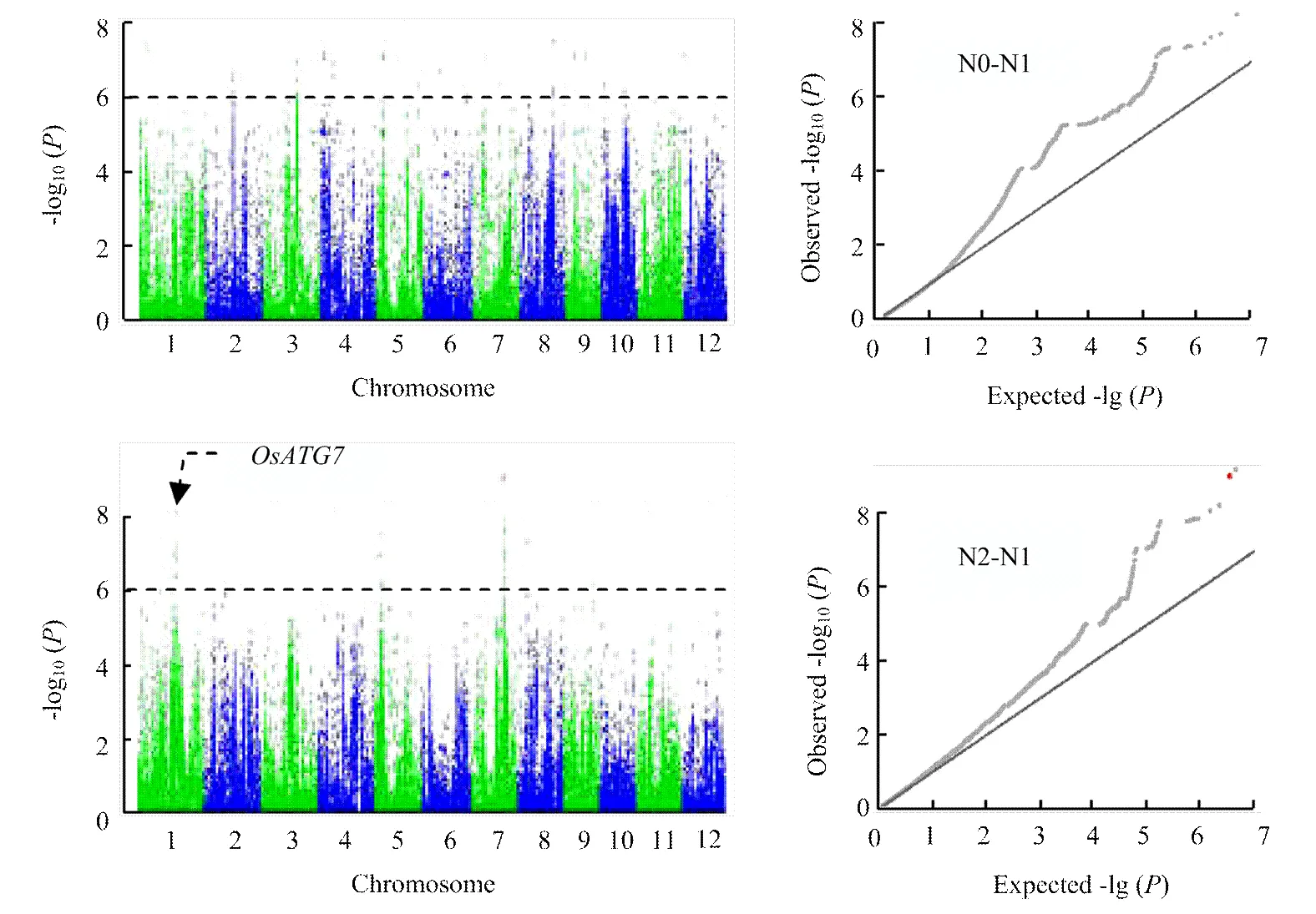
图6 剑叶叶宽对低氮和高氮差异响应的全基因组关联分析
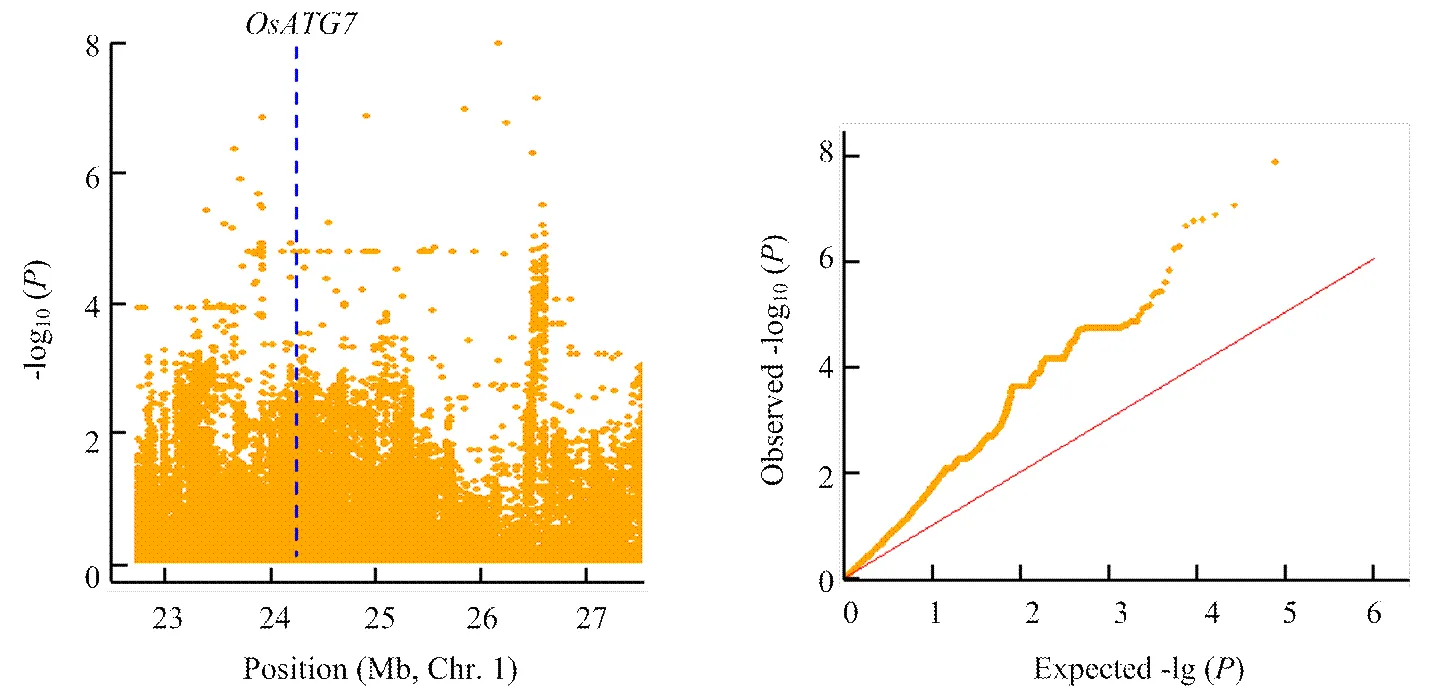
图7 第1染色体上在N2水平的关联显著位点候选区间分析
3 讨论
水稻成熟期剑叶形态是决定植株光合过程的受光面积和株型结构的关键因素,并进而影响灌浆期光合生物同化量的大小及水稻产量[36-37],因而改善水稻成熟期剑叶形态成为水稻保持优势株型和高产的重要手段。但水稻叶片形态是复杂的受微效多基因控制的数量性状,易受光、温、水、肥等环境影响[38]。近年来,随着GWAS在植物领域研究的应用,为发掘重要种质资源和快速鉴定新基因提供了新的分析工具[14,16]。
从GWAS的结果来看,水稻剑叶叶宽对氮肥胁迫的响应更为显著。在低氮或高氮水平下检测到的关联位点远多于正常氮肥氮肥水平下剑叶叶宽的关联位点,尤其以低氮响应为甚,这可能与剑叶叶宽易受氮肥水平的影响以及不同种质资源对低氮或高氮的响应方式不同有关。因而,对于易受环境影响的性状在开展GWAS分析时,最好能结合多环境下的分析或严格控制其他环境因素的影响。本研究采用3次重复的裂区设计,但没有作多年/异地的重复,可能会对部分关联位点的精准选择产生影响。在进行籼粳型的群体进行关联分析时,将BN (Balding-Nichols)矩阵纳入基于MLM模型的关联分析中,进行群体结构的校正,可以有效规避由于群体结构造成的影响,从而减少假阳性位点的产生。
氮素的同化和利用对植物至关重要,前人的研究结果来看,分别在第1、2、3、5、7、10、11、12染色体上存在已报道的与氮素相关QTL位点。其中,位于第2染色体的RM5812 标记与本研究的低氮胁迫下的检测到的显著性位点区间相近[39]。同样,在低氮胁迫下检测到的位于第3、第5染色体上的显著性位点所在区间也有QTL位点报道过(http://archive.gramene.org/)。因此,基于显著性的关联位点,需要进一步对候选区间内的基因进行鉴定、分析和克隆。后期的进一步表达水平变化或基因敲除研究对关联位点进行功能验证十分重要,深入展开关联候选基因的功能研究将有助于准确揭示水稻氮素利用遗传机制奠定基础。
4 结论
在不同氮肥水平下对水稻剑叶叶宽进行全基因组关联分析,共检测到的42个显著的关联位点中,其中14个与剑叶叶宽相关,28个与氮肥的响应相关。对所检测到的位点进行候选区间分析,发现2个位于候选区间内分别调控水稻剑叶叶宽及氮响应的已知基因;检测到多条染色体上的显著位点与已报道与氮素利用相关QTL区间相近。此外,基于候选区间基因的功能注释,检测到1个与已知调控叶宽的同源基因。
致谢:中国科学院遗传与发育生物学研究所田志喜研究员和李云海研究员为关联分析提供指导及关联群体材料,在此表示感谢。
References
[1] ANDREWS M, RAVEN J A, LEA P J. Do plants need nitrate? The mechanisms by which nitrogen form affects plants., 2013, 163: 174-199.
[2] ZHANG Q F. Strategies for developing green super rice., 2007, 104(42): 16402-16409.
[3] ZHANG Y J, TAN L B, ZHU Z F, YUAN L X, XIE D X, SUN C Q. TOND1 confers tolerance to nitrogen deficiency in rice., 2015, 81(3): 367-376.
[4] HU B, WANG W, OU S, TANG J, LI H, CHE R, ZHANG Z, CHAI X, WANG H, Wang Y, LIANG C, LIU L, PIAO Z, DENG Q, DENG K, XU C, LIANG Y, ZHANG L, LI L, CHU C. Variation in NRT1.1B contributes to nitrate-use divergence between rice subspecies., 2015, 47(7): 834-838.
[5] SUN H, QIAN Q, WU K, LUO J, WANG S, ZHANG C, MA Y, LIU Q, HUANG X, YUAN Q, HAN R, ZHAO M, DONG G, GUO L, ZHU X, GOU Z, WANG W, WU Y, LIN H, FU X. Heterotrimeric G proteins regulate nitrogen-use efficiency in rice., 2014, 46(2): 652.
[6] GALLAIS A, HIREL B. An approach to the genetics of nitrogen use efficiency in maize., 2004, 55(396): 295-306.
[7] HIREL B, LE GOUIS J, NEY B, GALLAIS A. The challenge of improving nitrogen use efficiency in crop plants: towards a more central role for genetic variability and quantitative genetics within integrated approaches., 2007, 58(9): 2369-2387.
[8] BI Y M, KANT S, CLARKE J, GIDDA S, MING F, XU J, ROCHON A, SHELP B J, HAO L, ZHAO R, MULLEN R T, ZHU T, ROTHSTEIN S J. Increased nitrogen-use efficiency in transgenic rice plants over-expressing a nitrogen-responsive early nodulin gene identified from rice expression profiling., 2009, 32(12):1749-1760.
[9] XU G, FAN X, MILLER A J. Plant nitrogen assimilation and use efficiency., 2012, 63(63): 153-182.
[10] TONG H H, MEI H W, YU X Q, XU X Y, LI M S, ZHANG SQ, LUO L J. Identification of related QTLs at late developmental stage in rice (L.) under two nitrogen levels., 2006, 33(5): 458-467.
[11] CHO Y I, JIANG W, CHIN J H, PIAO Z, CHO Y G, Mccouch S, Koh H J. Identification of QTLs associated with physiological nitrogen use efficiency in rice., 2007, 23(1): 72-79.
[12] 凌启鸿. 作物群体质量. 上海: 上海科技出版社, 2000: 42-120.
LING Q H.. Shanghai: Shanghai Scientific and Technical Publishers, 2000: 42-120. (in Chinese)
[13] 殷春渊, 王书玉, 薛应征, 刘贺梅, 张栩, 孙建权, 王和乐, 胡秀明. 氮肥处理对新稻18号水稻产量及叶片形态特征的影响. 中国农业科技导报, 2012, 14(3): 101-106.
YIN Q Y, WANG Y S, XUE Y Z, LIU H M, ZHANG X, SUN J Q, WANG H L, HU X M. Effects of nitrogen treatment on rice yield and leaves conformation characteristics of 'Xindao No.18'., 2012, 14(3): 101-106. (in Chinese)
[14] ZHAO K, TUNG C W, EIZENGA G C, WRIGHT M H, ALI M L, PRICE A H, NORTON G J, ISLAM M R, REYNOLDS A, MEZEY J, MCCLUNG A M, BUSTAMANTE C D, MCCOUCH S R. Genome-wide association mapping reveals a rich genetic architecture of complex traits in., 2011, 2(1): 1020-1021.
[15] ATWELL S, HUANG YS, VILHJÁLMSSON BJ, WILLEMS G, HORTON M, LI Y, MENG D, PLATT A, TARONE A M, HU T T, JIANG R, MULIYATI N W, ZHANG X, AMER M A, BAXTER I, BRACHI B, CHORY J, DEAN C, DEBIEU M, DE MEAUX J, ECKER JR, FAURE N, KNISKERN JM, JONES JD, MICHAEL T, NEMRI A, ROUX F, SALT DE, TANG C, TODESCO M, TRAW MB, WEIGEL D, MARJORAM P, BOREVITZ JO, BERGELSON J, NORDBORG M. Genome-wide association study of 107 phenotypes ininbred lines., 2010, 465(7298): 627-631.
[16] HUANG X H, WEI X H, SANG T, ZHAO Q, FENG Q, ZHAO Y, LI C, ZHU C, LU T, ZHANG Z, LI M, FAN D, GUO Y, WANG A, WANG L, DENG L, LI W, LU Y, WENG Q, LIU K, HUANG T, ZHOU T, JING Y, LI W, LIN Z, BUCKLER ES, QIAN Q, ZHANG Q F, LI J, HAN B. Genome-wide association studies of 14 agronomic traits in rice landraces., 2010, 42(11): 961-967.
[17] ZHOU Z K, JIANG Y, WANG Z, GOU Z, LYU J, LI W, YU Y, SHU L, ZHAO Y, MA Y, FANG C, SHEN Y, LIU T, LI C, LI Q, WU M, WANG M, WU Y, DONG Y, WAN W, WANG X, DING Z, GAO Y, XIANG H, ZHU B, LEE S H, WANG W, TIAN Z X. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean., 2015, 33(4): 408-414.
[18] WEN W, LI D, LI X, GAO Y, LI W, LI H, LIU J, LIU H, CHEN W, LUO J, YAN J. Metabolome-based genome-wide association study of maize kernel leads to novel biochemical insights., 2015, 5(2): 487-507.
[19] KUMAR V, SINGH A, MITHRA S V, KRISHNAMURTHY S L, PARIDA S K, JAIN S, TIWARI K K, KUMAR P, RAO A R, SHARMA S K, KHURANA J P, SINGH N K, MOHAPATRA T.Genome-wide association mapping of salinity tolerance in rice ()., 2015, 22(2): 133-145.
[20] WANG C, YANG Y, YUAN X, XU Q, FENG Y, YU H, WANG Y, WEI X. Genome-wide association study of blast resistance in indica rice., 2014, 14(1): 1-11.
[21] ZHU D, KANG H, LI Z, LIU M, ZHU X, WANG Y, WANG D, WANG Z, LIU W, WANG G L. A genome-wide association study of field resistance to magnaporthein rice., 2016, 9(1): 44.
[22] CHEN W, GAO Y, XIE W, GONG L, LU K, WANG W, LI Y, LIU X, ZHANG H, DONG H, ZHANG W, ZHANG L, YU S, WANG G, LIAN X, LUO J. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism., 2014, 46(7):714-721.
[23] 王兰, 黄李超, 代丽萍, 杨窑龙, 徐杰, 冷语佳, 张光恒, 胡江, 朱丽, 高振宇, 董国军, 郭龙彪, 钱前, 曾大力. 利用日本晴/9311重组自交系群体定位水稻成熟期叶形相关性状QTL. 中国水稻科学, 2014, 28(6): 589-597.
WANG L, HUANG L C, DAI L P, YANG Y L, XUE J, LENG Y J, ZHANG G H, HU J, GAO Z Y, DONG G J, GUO L B, QIAN Q, ZENG D L. QTL analysis for rice leaf morphology at maturity stage using a recombinant inbred line population derived from a cross between nipponbare and 9311., 2014, 28(6): 589-597. (in Chinese)
[24] Murray M G, Thompson W F. Rapid isolation of high molecular weight plant DNA., 1980, 8(19): 4321-4325.
[25] Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data., 2010, 38(16): e164.
[26] Felsenstein J.PHYLIP-phylogeny inference package (version 3.2)., 1989, 5: 164-166.
[27] Price A L, Patterson N J, Plenge R M, Weinblatt M E, Shadick N A, Reich D. Principal components analysis corrects for stratification in genome-wide association studies., 2006, 38(8): 904-909.
[28] Kang H M, Sul J H, Service S K, Zaitlen N A, Kong S Y, Freimer N B, Sabatti C, Eskin E. Variance component model to account for sample structure in genome-wide association studies., 2010, 42(4): 348-354.
[29] Dudbridge F, Gusnanto A. Estimation of significance thresholds for genomewide association scans., 2008, 32(3): 227-234.
[30] MORI M, NOMURA T, OOKA H, ISHIZAKA M, YOKOTA T, SUGIMOTO K, OKABE K, KAJIWARA H, SATOH K, YAMAMOTO K, HIROCHIKA H, KIKUCHI S. Isolation and characterization of a rice dwarf mutant with a defect in brassinosteroid biosynthesis., 2002, 130(3): 1152-1161.
[31] HONG Z, UEGUCHI-TANAKA M, SHIMIZU-SATO S, INUKAI Y, FUJIOKA S, SHIMADA Y, TAKATSUTO S, AGETSUMA M, YOSHIDA S, WATANABE Y, UOZU S, KITANO H, ASHIKARI M, MATSUOKA M. Loss-of-function of a rice brassinosteroid biosynthetic enzyme, C-6 oxidase, prevents the organized arrangement and polar elongation of cells in the leaves and stem., 2002, 32(4): 495-508.
[32] LI M, XIONG G, LI R, CUI J, TANG D, ZHANG B, PAULY M, CHENG Z, ZHOU Y. Rice cellulose synthase-like D4 is essential for normal cell-wall biosynthesis and plant growth., 2009, 60(6):1055-1069.
[33] HU J, ZHU L, ZENG D, GAO Z, GUO L, FANG Y, ZHANG G, DONG G, YAN M, LIU J, QIAN Q. Identification and characterization of NARROW AND ROLLED LEAF 1, a novel gene regulating leaf morphology and plant architecture in rice., 2010, 73(3): 283-292.
[34] WADA S, HAYASHIDA Y, IZUMI M, KURUSU T, HANAMATA S, KANNO K, KOJIMA S, YAMAYA T, KUCHITSU K, MAKINO A, ISHIDA H. Autophagy supports biomass production and nitrogen use efficiency at the vegetative stage in rice., 2015, 168(1): 60-73.
[35] KURUSU T, KOYANO T, HANAMATA S, KUBO T, NOGUCHI Y, YAGI C, NAGATA N, YAMAMOTO T, OHNISHI T, OKAZAKI Y, KITAHATA N, ANDO D, ISHIKAWA M, WADA S, MIYAO A, HIROCHIKA H, SHIMADA H, MAKINO A, SAITO K, ISHIDA H, KINOSHITA T, KURATA N, KUCHITSU K. OsATG7 is required for autophagy-dependent lipid metabolism in rice postmeiotic anther development., 2014, 10(5): 878-888.
[36] LI Z K, PINSON S R, STANSEL J W, PATERSON A H. Genetic dissection of the source-sink relationship affecting fecundity and yield in rice (L.)., 1998, 4: 419-426.
[37] YUE B, XUE W Y, LUO L J, XING Y Z. QTL Analysis for flag leaf characteristics and their relationships with yield and yield traits in rice.2006, 33: 824-832.
[38] ZHU Y, CHANG L, TANG L, JIANG H, ZHANG W, CAO W. Modelling leaf shape dynamics in rice., 2009, 57(1): 73-81.
[39] Dai G J, Cheng S H, Hua Z T, Zhang M L, Jiang H B, Feng Y, Shen X H, Su Y A, He N, Ma Z B, Ma X Q, Hou S G, Wang Y R. Mapping quantitative trait loci for nitrogen uptake and utilization efficiency in rice (L.) at different nitrogen fertilizer levels., 2015, 14(3): 10404-10414.
(责任编辑 李莉)
Genome-wide association analysis on flag leaf width under different nitrogenlevels in rice
GAO YiHong, YAN JinXiang, TU ZhengJun, LENG YuJia, CHEN Long, HUANG LiChao, DAI LiPing, ZHANG GuangHeng, ZHU Li, HU Jiang, REN DeYong, GUO LongBiao, QIAN Qian, WANG DanYing, ZENG DaLi
(China National Rice Research Institute/State Key Laboratory of Rice Biology, HangZhou 310006)
【Objective】 The objective of this experiment is to study the genetic mechanism of flag leaf width and its response to different nitrogen fertilizer rates, and provide advantageous germplasm resources and genetic markers for the improvement of nitrogen use efficiency in rice breeding. 【Method】Based on re-sequencing of 134 rice landraces, a total of 3 356 591 SNPs distributed on the whole genome were identified. Three different nitrogen levels were assigned as the main plot in the split-plot design, and rice landraces are assigned at random to the subplots within each whole plot. Three nitrogen levels including low nitrogen (no nitrogen fertilizer), normal nitrogen (96 kg·hm-2) and high nitrogen (192 kg·hm-2) were applied under normal field cultivation, respectively. The EMMAX method was used to analyze the genetic relationship and EIGENSOFT was employed to detect the population structure. The mixed linear model was used to detect the potential genome-wide association between single nucleotide polymorphisms (SNPs) and the flag leaf width performance or response under low, medium and high nitrogen treatments. 【Result】The results showed that the flag leaf width displayed normal distribution in N0, N1 and N2 treatments, respectively. The variation of flag leaf width caused by the varietal differences and the different nitrogen levels, and the significant positive correlation between nitrogen fertilizer and flag leaf width were detected at all nitrogen levels. A total of 14 SNPs on five chromosomes presented significant association with the flag leaf width under three different nitrogen levels. The estimated minimum allele frequency was 0.46 at low nitrogen level, which indicate those loci are widely distributed in the association population. While the estimated minimum allele frequencies were lower at normal nitrogen and high nitrogen levels. One SNP located on chromosome 12 was found both at normal nitrogen and high nitrogen levels. Besides, another SNP on chromosome 12 was also detected at high nitrogen. Its flank contained a candidate gene, which belongs to the super-family of cytochrome P450. It is a homologous gene ofwhich was confirmed inregulating the flag leaf width in rice. Based on the variation of leaf width at different nitrogen levels, twenty SNPs and eight SNPs were identified to low nitrogen and high nitrogen response, respectively. Among them, the nitrogen utilization related gene,, was associated with high nitrogen response on chromosome 1. 【Conclusion】In this study, a total of 42 SNPs associated with flag leaf width and response to different nitrogen were identified based on the genome-wide association analysis.
; genome-wide association study; leaf width; nitrogen fertilizer
2017-01-12;接受日期:2017-04-05
国家自然科学基金(91435105、31661143006、31371581)、中国农业科学院科技创新工程项目
高易宏,E-mail:gaoyihong93@163.com。燕金香,E-mail:2911669220@qq.com。高易宏和燕金香为同等贡献作者。通信作者曾大力,E-mail:dalizeng@126.com。通信作者王丹英,E-mail:wangdanying@caas.cn
猜你喜欢
中国化肥信息(2022年9期)2022-11-23
中国水稻科学(2021年6期)2021-11-18
新疆农业科学(2020年1期)2020-02-14
河南农业科学(2019年12期)2019-12-05
中国化肥信息(2019年4期)2019-05-31
中国化肥信息(2019年3期)2019-04-25
浙江农业科学(2019年1期)2019-01-25
江苏农业学报(2018年5期)2018-09-10
中国化肥信息(2017年3期)2017-12-23
江苏农业科学(2016年10期)2017-02-05
