
气相中Ni+活化环己烷C—H和C—C键的理论研究
2018-01-01 21:35袁永宁刘瑞瑞耿志远
科学与财富 2017年30期
袁永宁+刘瑞瑞+耿志远
摘要:使用密度泛函理论(DFT)详细研究了二,四重态势能面(PES)上过渡金属Ni+活化环己烷中C-H和C-C键的反应机理.计算结果表明:Ni+与环己烷的反应只在二重态势能面上进行,无势能面交叉.Ni+独特的电子结构可以活化环己烷中的C-H和C-C键.C-H键断裂,伴有副产物H2产生.C-C键断裂,会发生结构翻转,Ni从环己烯的一面翻转至另一面,伴有副产物D2的形成,最终生成化合物苯.
关键词:密度泛函理论;C-H和C-C键活化;结构翻转
1.引言
金属有机催化因其在合成转化中的广泛应用,不论在工业界还是学术界都受到了广泛的关注.尤其是Ni等过渡金属可以选择性的活化碳氢化合物中C-H和C-C键,这在有机化学,生物化学,石油工业,表面化学和催化研究方面有至关重要的作用。
2.计算方法
本文使用密度泛函理论中的B3LYP方法对二重态和四重态势能面上所有的反应物,产物,中间体,过渡态的几何构型进行了优化计算.氢和碳元素采用6-31G**基组,过渡金属Ni采用LANL2DZ基组对每个固定结构进行振动频率计算,以确定它是中间体还是过渡态(TS),并通过内禀反应坐标(IRC)计算,进一步确认过渡态正确连接两个中间体.这些计算都是在高斯09程序下进行的[21].
3.结果与讨论
3.1 C-H键的活化过程
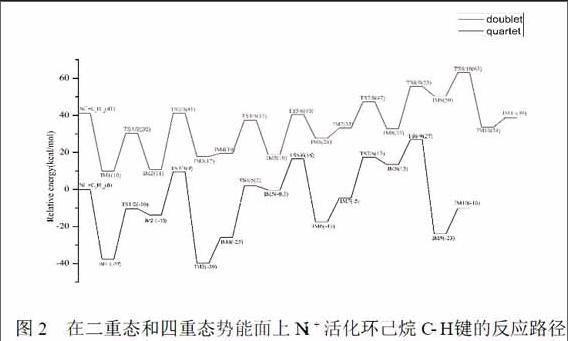
在Ni+活化环己烷的反应中,为了确定不同势能面间是否会发生自旋翻转,我们对两个自旋态下的反应进行了研究.计算结果表明,Ni+的基态是二重态(2D,3d9),其能量比第一激发态(4F,3d84s1)低41kcal/mol;另一方面,Ni+不论是活化环己烷中的C-H键还是C-C键,反应均只在二重态势能面上进行,无势能面交叉.所以,在本文中我们只研究发生在二重态势能面上的反应,对四重态不做讨论.
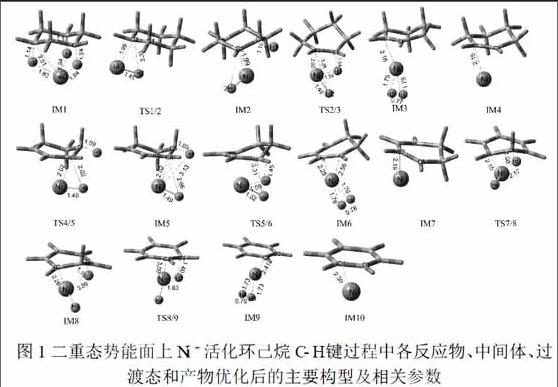
3.1.1 第一分子氢气的形成过程.如图1所示,环己烷分子与2Ni+相互作用形成初始复合物IM1,二者的配体结合能为-37kcal/mol.在过渡态TS1/2中,由于Ni催化环己烷的区域选择性,过渡金属Ni中心靠近C2(Ni-C2键长从2.51缩短至1.99),而C2-H2键从1.14A延伸至2.39 ,
该异构化表明,Ni-C之间的相互作用有效的促进了C-H键断裂,从而加速了氢原子的迁移.通过TS2/3,C2-H2键长从2.39 延伸至3.28 ,C1-H1键长从1.10 延伸至1.54 ,表明C1-H1和C2-H2键同时断裂.随着C-H键的彻底断裂,加之Ni+与两个氢原子的作用力变强,至使Ni+携带两个H原子脱离环己烷而逸出.在IM3中,H1-H2距离从1.71 变短到0.77 ,这个特征表明H1-H2键已完全形成,氢气生成(氢分子距离为0.70 ).第一分子氢气消除的整个过程中,最高的活化能仅为9kcal/mol,表明第一分子H2的形成反应很容易发生.
3.1.2第二,三分子氢气的形成过程.在形成中间体IM4后,反应经历第三个C-H键的断裂.在TS4/5中,形成了等腰平面的C6-Ni-H6复合物,其中Ni原子接近环己烯中心,C6-H6距离随着Ni的靠近而稍微拉长.在IM5中,H6远离碳环中心形成C6-Ni-H6三元环配位键,使得碳环异构化,从而增强了Ni和另外一个H原子的作用力,所以,通过TS5/6,C5-H5键从1.09 延伸至1.45 ,表明C5-H5键完全断裂.下一步是H2的形成,在IM5中,H5-H6间的距离等于0.78 .表明H5-H6共价键形成,最终解离成H2分子.第二分子氢气形成需吸热16kcal/mol的能量.
第三分子氢气的形成过程是从IM7开始到IM10结束.其中TS7/8到TS8/9是C-H键的断裂和H原子的迁移,两个过程同时发生.我们看到IM7是一个通过H2的还原消除而产生的具有不饱和键(C1=C2和C5=C6)的特殊结构,这些不饱和键将成为Ni插入到环己二烯形成新的C-Ni键的活性位点,这为C4-H4键的弱化和断裂起到重要作用,同时也表明sp2→sp3杂化对H2形成是有利的.三分子氢气都脱去形成的最终产物IM10是一个Ni-η6-苯的六配位络合物.
3.2 C-C键的活化过程
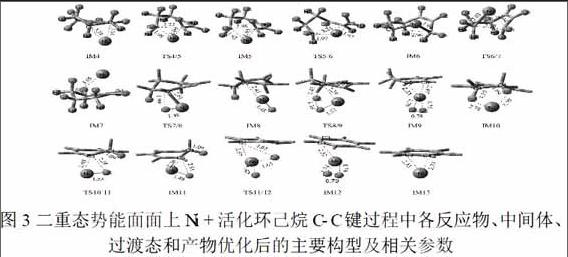
根据图2中的势能面,我们发现Ni+优先活化环己烷中的C-H键,脱氢形成环己烯/Ni+后,多次脱氢过程变的困难.此时,出现两种竞争机制,一种是Ni+继续活化C-H键形成Ni(C6H10-nD6,n=1-4)+络合物,该过程与第一分子氢气形成相似.另一种是Ni+在活化C-H键的同时也活化C-C键,得到Ni(C6H10-nDn,n=2-4)+络合物.Ni+在二重态势能面上活化环己烯的C-C键过程中各反应物、中间体、过渡态和产物优化后的主要构型及相关参数表示在图3中,在二重态和四重态势能面上Ni+活化环己烷C-C键的反应路径表示在图4中.通过对这些优化好的结构的观察,我们发现对于一分子D2的形成,主要经过C-C单键裂解;断键后结构的空间旋转和闭环反应;C-D键的断裂和D2的还原消除.
惰性C(sp3)—C(sp3)单键的断裂,需要很高能量. 所以,从热力学上来说,C-C键断裂是受阻的.从动力学上考虑,我们发现C-Cσ键有很强的方向性,它是沿着键轴方向的.要想发生显著变形需要它的前线分子轨道和金属轨道的有效重叠,而断键过渡态TS3/4的前线分子轨道的最高占据轨道(HOMO)是由Ni的3dyz和C的pxy形成的反键轨道,最低空轨道(LUMO)则是由Ni的3dz2和C的p轨道杂化形成的成键轨道.这种有效重叠,可以使C-Cσ键减轻了成键轨道/反键轨道的方向性.另一方面,也导致反应底物环己烯不稳定,有助于降低活化屏障,最终使得C3-C4键断裂,环己烯环打开.通过打开环己烯环可以减小空间位阻从而形成张力较小的七元环IM5(图3),这可以为Ni+反转到另一面提供驱动力,尽管这种促进力很小.通过图2和图4势能面的比较,我们发现在第二分子H2形成过程中两个C-H键活化(2kal/mol,16kal/mol)所需能量比第一分子D2形成过程中的C-C键活化(10kal/mol)能量高,这充分表明在同等条件下,相同反应物中C-C键活化和C-H键活化存在很强的竞争效应.
环己烯C3-C4键断裂后,为了使Ni+翻转至环己烯另一面,我们提出一个环反转机理.金属Ni+通过改变配位点插入环己烯C-C键,得到IM5/Ni+(图3).随后,IM5/Ni+可以进行环转换(“翻转”),经过TS5/6这个过渡态后形成IM6/Ni+,最后C-C键重新闭合,镍离子进入到另一表面并形成环己烯反式异构体IM7/Ni+.通过这种方法,我们可以实现简单的H/D平衡过程.此外,伴隨着Ni+和环己烯C=C双键的平面配位,Ni+空的3d轨道可以允许IM4/Ni+ IM7/Ni+的直接相互转换.
C-D键断裂后所形成D2的过程和H2的还原消除过程相似,在允许计算误差存在的情况下,所需能量也大致相同:第一分子D2(所需最高能量17kcal/mol)和第二分子H2(所需最高能量16kcal/mol);第二分子D2(所需最高能量27kcal/mol)和第三分子H2(所需最高能量27kcal/mol)所需能量一样高,在热力学上是不能发生的,这也和实验结果相一致.
4结论
我们运用密度泛函理论(DFT)中b3lyp方法对气相中Ni+活化环己烷中C-H和C-C键的反应进行了理论研究.通过对反应路径的分析和比较,我们总结出了以下结论.首先,详细的零点势能证明,在一分子H2或者D2形成过程中,断裂的前一个C-H键或者C-D键均比第二个所需的能量低.相似的,后一分子H2或者D2都比前一分子难形成.第三分子H2和第二分子D2是最难形成的,所需要的能量高达27kcal/mol,这在室温下是难以发生的,和实验结果相吻合.不论是活化C-H键还是C-C键,整个反应过程只在二重态势能面上发生,无势能面交叉.体系的计算结果对深入认识过渡金属离子催化碳氢化合物的过程具有一定的学术意义.endprint
