
SIR-DNA传染病动力学优化算法*
2018-02-05 03:46陆秋琴黄光球
计算机与生活 2018年2期
陆秋琴,黄光球
西安建筑科技大学 管理学院,西安 710055
1 引言
DNA计算[1]是利用DNA双螺旋结构和碱基互补配对规律进行信息编码,将要运算的对象映射成DNA分子链,通过生物酶的作用,生成各种数据池,再按照一定的规则将原始问题的数据运算高度并行地映射成DNA分子链的可控的生化反应过程,然后利用分子生物技术,检测所需要的运算结果[2]。
DNA计算包括分子内DNA计算[3]、分子间DNA计算[1]和超分子DNA计算[2]。分子内DNA计算借助于分子内的形态转移操作,用单DNA分子构建可编程的状态机[3];分子间DNA计算集中在不同DNA分子间的杂交反应,使其作为计算中的一个基本步骤[1];超分子DNA计算是利用不同序列的原始DNA分子的自装配过程进行计算[4-5]。
DNA计算利用DNA反应的强大并行计算能力,已成功地解决了许多NP难题。吴帆等人[4]提出了一种基于DNA自组装模型来求解N皇后问题的DNA计算方法,该算法降低了生化解的错误率。李肯立等人[5]通过引入DNA自组装模型,提出了一种求解最大团问题的DNA计算方法,该算法给出了DNA分子的编码方案及结果检测的实验方法。姚庆安等人[6]提出一种基于k-臂分子和粘贴计算求解最短路径问题的DNA计算模型。徐京雷等人[7]提出了一种基于改进的闭环模型求解TSP问题的DNA算法,该算法能在较低的时间复杂度内解决TSP问题。马莹等人[8]将着色问题转化为求最大独立集问题,然后通过质粒DNA分子生物实验得到最大独立集,证明了该质粒DNA算法有效并且是可行的。吴雪等人[9]针对最大匹配问题,应用DNA粘贴计算模型寻求该问题最优解的生物操作过程,实例表明该算法是可行的。
目前,DNA计算的大量研究还停留在纸面上,很多设想和方案没有条件付诸实验[2],对维数较高的优化问题的求解还存在一些困难。为解决此问题,本文从一个新的角度提出了一种新的DNA算法。本文算法依据一种传染病在一个生态系统中传播的场景构造而得,该生态系统中动物被感染后,传染病攻击该动物的某些DNA片断,从而导致该动物具有类似遗传病的缺陷。本文算法不需依赖生物实验即可进行优化计算。
在现实世界,人群中经常爆发的很多传染病均可用KM仓室建模方法[10-11]描述。传染病在个体之间的传播,使得个体之间的相互作用关系体现得淋漓尽致;一种传染病攻击的是人类个体的少部分器官,将该现象映射到对优化问题的求解,就是每次处理的变量个数只是全部变量的极少部分。因此,该算法具有求解高维问题的天然优势。为简单起见,本文利用SIR疾病传播场景来说明本文的思路,所提出的优化算法称为SIR-DNA算法。
2 SIR-DNA算法设计
考虑优化问题:
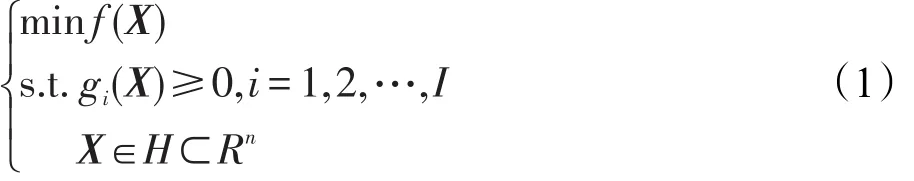
式中,Rn是n维欧氏空间;X=(x1,x2,…,xn)是一个n维决策向量;H为搜索空间,又称解空间;f(X)为目标函数;gi(X)≥0为第i个不等式约束条件,i=1,2,…,I,I为不等式约束条件个数。目标函数f(X)和约束条件gi(X)不需要特殊的限制条件。
把优化模型式(1)中约束条件所形成的可行解空间设想成一个生态系统ES。假设ES包含有N个动物,每个动物称为一个个体,这些个体的编号是1,2,…,N。有一种传染病Z在该生态系统中传播,动物被感染后,传染病Z攻击该动物的某些DNA片断,从而导致该动物具有类似遗传病的缺陷。在现实世界,HIV、天花、SARS、寨卡等传染病毒就是这样的病毒。
2.1 动物个体DNA分子
动物的每条染色体携带一个DNA分子,DNA是由分别带有4种碱基A、T、C、G的脱氧核苷酸链接组成的双螺旋长链分子。在这条双螺旋的长链中,共有约30亿个碱基对,而基因则是DNA长链中有遗传效应的一些片段。在组成DNA的数量浩瀚的碱基对中,有一些特定位置的单个核苷酸经常发生变异引起DNA的多态性,人们称之为位点。染色体、基因和位点的结构关系如图1所示。在DNA长链中,位点个数约为碱基对个数的1/1 000。由于位点在DNA长链中出现频繁,多态性丰富,近年来成为人们研究DNA遗传信息的重要载体。大量研究表明,动物的许多表型性状差异以及对传染病毒的易感性等都可能与某些位点相关联,或和包含有多个位点的基因相关联。
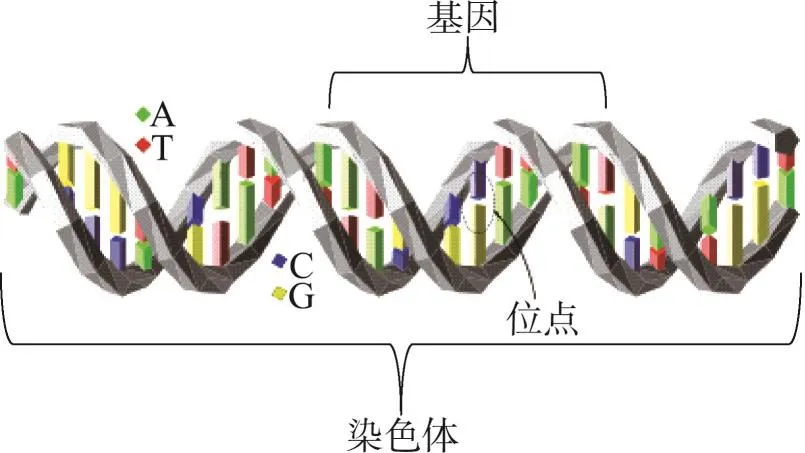
Fig.1 Structure of chromosomes,genes and loci图1 染色体、基因和位点的结构关系
对于ES中的每个动物个体,采用A、T、C、G的编码方式来获取每个位点的信息,因为染色体具有双螺旋结构,所以用两个碱基的组合表示一个位点的信息,如表1所示。表1中,在位点rs100015位置,不同样本的编码都是T和C的组合,有3种不同编码方式TT、TC和CC。类似地,其他位点虽然碱基的组合不同,但也只有3种不同编码。
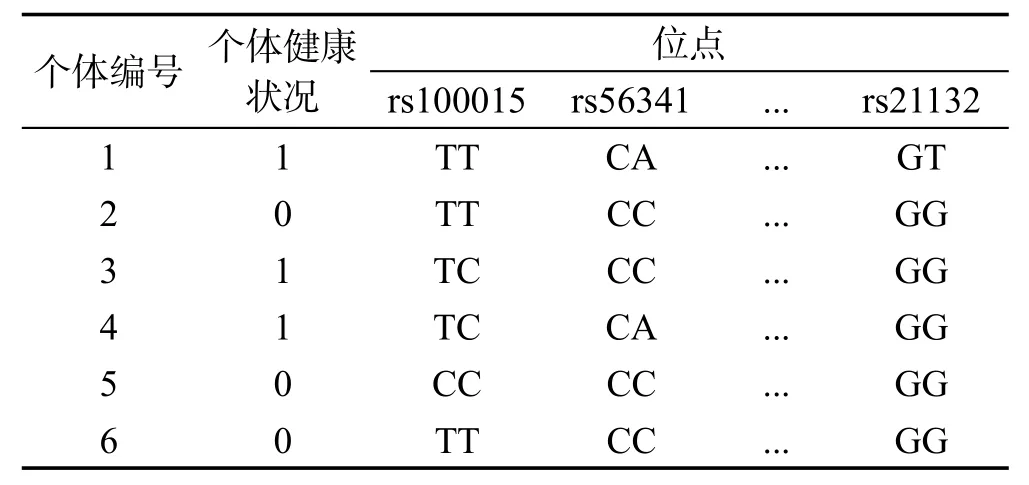
Table 1 Loci name and loci alleles of DNAfragments表1 DNA片段位点名称和位点等位基因信息
在ES中,传染病Z攻击的是动物个体W个特殊基因中某些位点,这些特殊基因称为致病基因,而这W个致病基因均由R个位点组成,其编号为1,2,…,R;每个位点均由3个碱基对组成。对于不同的动物个体,传染病Z攻击的是W个致病基因中的哪些基因完全是随机的;若某个致病基因被攻击,组成该基因的R个位点中的哪些位点受到攻击也完全是随机的。染病个体治愈后会获得短期免疫力,与免疫相关的特殊基因也有W个,这些特殊基因称为免疫基因,每个免疫基因也包含R个位点;愈后个体在哪些免疫基因中的哪些位点获得免疫,也是完全随机的;虽然个体在不同免疫基因的不同位点上获得免疫力,但是免疫效果都是一样。为计算处理方便,将A、T、C、G采用二进制编码分别表示为00、01、10、11。
2.2 SIR模型转换成个体类别转换方程
将ES中的动物群分成易感者类(S)、染病者类(I)和治愈者类(R)。易感个体一旦与染病者接触,就会因感染而变成染病者;染病者经过治疗后会成为治愈者;治愈者能够获得暂时的免疫,但免疫丧失后又会变成易感者。假设该传染病不是恶性的,故在一个时间段内,该传染病不会造成动物死亡;此外,不考虑该时间段内个体的出生、死亡、迁入和迁出。故可假设该ES中的动物个体数N为常数。若传染病的发生率是饱和接触率,则基于KM经典的假设[10-11],该生态系统可用SIR传染病模型描述[12],如图2所示,其数学模型为式(2)所示。
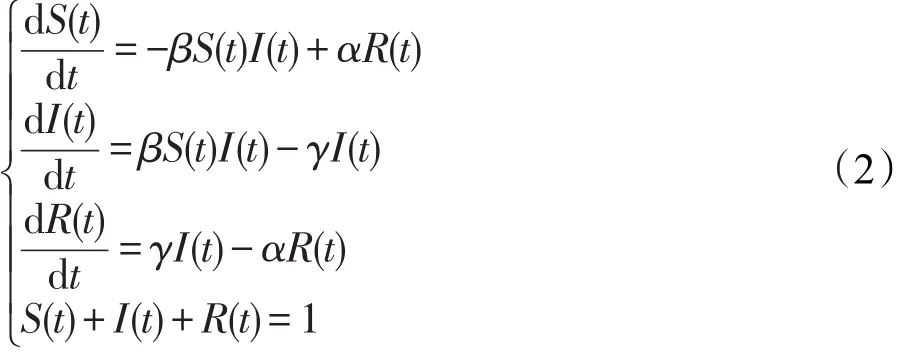
式中,β为动物种群饱和接触传染率;γ为治愈系数;α为免疫丧失率;S(t)、I(t)、R(t)分别为时期t易感者、染病者和治愈者的占比。

Fig.2 Bin structure of SIR epidemic model图2 SIR传染病仓室结构
因S(t)+I(t)+R(t)=1,S(t)、I(t)和R(t)实际上描述了一个概率分布,故对任意一个个体来说,S(t)、I(t)、R(t)相当于该个体属于易感者类、染病者类和治愈者类的概率,不妨将其分别称为易感概率、染病概率和治愈概率。因此,式(2)实际上是计算任意一个个体在时期t的属于易感者类、染病者类和治愈者类的概率的方程。为快速计算起见,将式(2)写成递推形式,并注意到式(2)中有一个方程是多余的,可以去掉,故对于个体i来说,有:
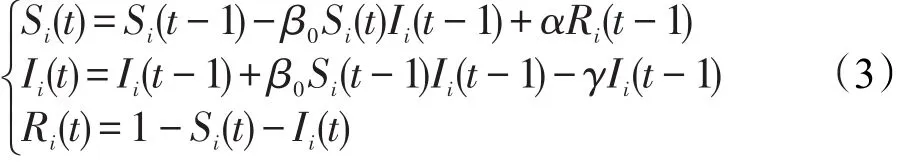
在时期t,采用式(3)计算出个体i的Si(t)、Ii(t)和Ri(t)。显然,在某个时期,任何一个个体只能处于S类、I类和R类中的某一个类。于是,个体i在时期t处于S类、I类和R类3个类别中的哪个类,可由Si(t)、Ii(t)和Ri(t)所决定的概率分布确定。表2中所列情况符合图2描述的SIR模型的类别转换情形。

Table 2 Legal class transfer of SIR epidemic model表2 SIR传染病模型的合法类别转换
既然优化问题式(1)的可行解空间与ES相对应,那么该ES中一个个体对应于优化问题式(1)的一个可行解,N个个体所对应的可行解集就是:

个体i的特征j对应于可行解Xi中的一个变量xij,j=1,2,…,K+n。因此,个体i与可行解Xi是等价概念。个体的体质强弱用体质指数IPI来表示,IPI指数对应于优化问题式(1)的目标函数值。好的可行解对应具有较高IPI值的个体,即体质强壮的个体,差的可行解对应具有较低IPI值的个体,即体质虚弱的个体。个体i的IPI指数计算方法为:

2.3 基因表达设计
在ES中,因动物个体的种类相同,故对于不同的个体,传染病毒Z攻击的W个特殊基因是相似的(如图3(a)所示),但不会相同,该特殊基因组称为致病基因组;这W个致病基因被攻击后发生改变,就会使动物患病。类似地,对于同种动物个体,能够对传染病毒Z免疫的W个免疫基因是类似的,但不会相同,该基因组称为免疫基因组,如图3(b)所示;对于同种动物中的不同个体,传染病被治愈后,这W个免疫基因会发生变化,会使动物获得免疫。免疫基因组和致病基因组是不同序列的基因组。
因动物个体之间存在差异,故传染病毒攻击致病基因组中的哪些致病基因,以及若一个致病基因被攻击,则该致病基因中的哪些位点发生改变,则完全因人而异。同理,当动物个体染病后被治愈并获得短期免疫力,但该个体在免疫基因组中的哪些免疫基因,以及若一个免疫基因获得免疫力,则该免疫基因中的哪些位点发生改变,完全因人而异。
传染病毒Z攻击动物个体的致病基因组和免疫基因组序列分别为:
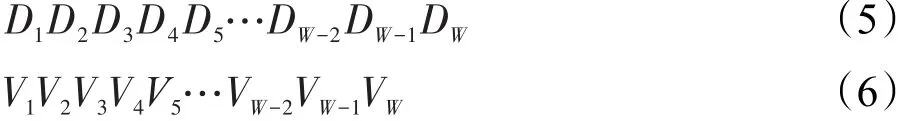
式(5)、(6)中的基因Di、Vi,i=1,2,…,W,均由R个位点组成,即:
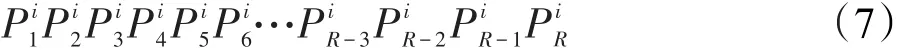
因每个动物个体是不相同的,故组成每个基因的位点是不相同的,从而对不同的动物个体来说,式(7)中的取值是不相同的,每个位点的取值均是从A、T、C、G中任取两个进行组合。例如,假设X和Y是某个位点的碱基对,则可能的组合只有XX、XY、YY3个。若用二进制表示,则每个基因的总长度是12R个比特,而W个基因序列的总长度是12RW个比特。
2.4 基因表达与变量的对应关系设计
模型式(1)中的未知数变量有n+K个,其中n=|H|为实数变量个数,K=|D|为0、1整数变量个数。显然,取W=n+K。于是,W个基因被传染病毒Z攻击,等价于n+K个未知数变量被处理。特别注意,传染病毒Z每次只选择几个基因实施攻击,此意味着每次只需处理n+K个未知数变量中的几个变量,于是达到了天然降维的目的。
W个致病基因被攻击后发生改变,在外显特征上,动物个体会表现出一些奇特的特征,如艾滋病人的免疫丧失,麻风病人脸上出天花,SARS病人呼吸窘迫等。然而,在SIR-DNA算法中,将动物个体的外显特征解释为个体性状,而一种个体性状对应着模型式(1)的一个可行解。由所有个体性状组成的表称为个体性状表,如图4(a)所示。

Fig.3 Gene expression before and after immunization and individual animals are infected图3 动物个体被传染病毒攻击前后和获得免疫前后的基因表达
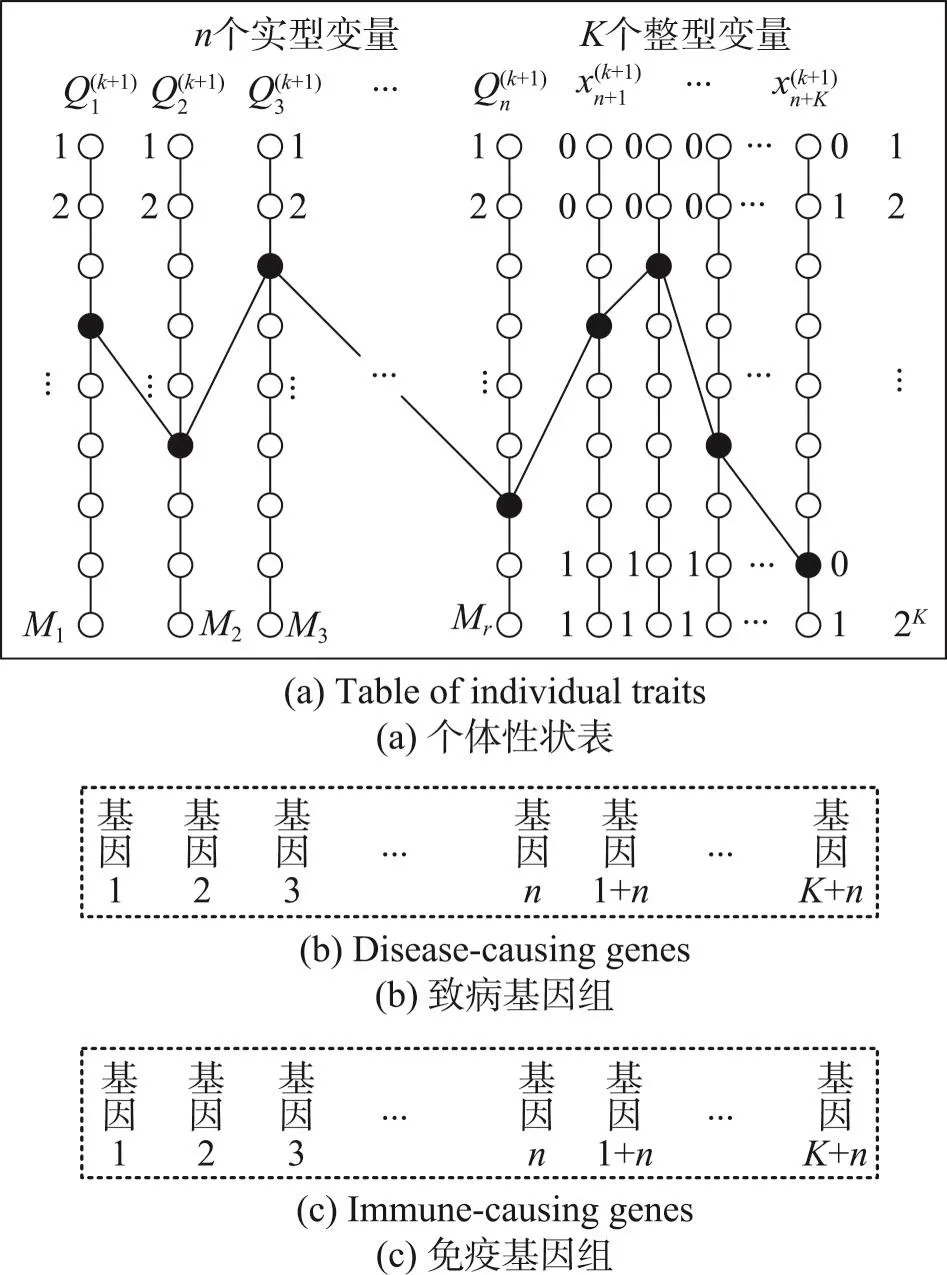
Fig.4 Corresponding relation between W disease-causing and immune-causing genes and W variables图4 W个致病基因和免疫基因与W个变量的对应关系
从外显特征看,若一个动物个体染上传染病Z,也获得对传染病Z的免疫,该个体属于S类;若一个动物个体染上传染病Z,即其致病基因发生改变,该个体属于I类;若一个动物个体染上传染病Z后被治愈,即其致病基因复原,其免疫基因发生改变,该个体属于R类。
图4描述了W个致病基因(如图4(b)所示)、W个免疫基因(如图4(c)所示)与W个变量的对应关系。图4(a)中,每个圆点代表一个位点;前n个为基因对应n个实数型变量;后K个为基因对应K个0-1整数型变量。每个基因上选择一个圆点,并组成一种个体性状,它对应着动物个体的一种外显特征。在本算法中,一种个体性状对应着一个可行解。例如,图中的所有黑点组成一个可行解。于是,对模型式(1)的求解最终转化为在图4(a)所示的各种离散点组合中,找出使模型式(1)的目标函数值达到最小的可行解,此为要寻找的全局最优可行解。
当传染病毒Z发起对动物个体的攻击时,因为该传染病毒每次只选择几个基因实施破坏,例如图4中传染病毒Z在一次攻击中只选择基因1、基因2和基因n+1实施破坏,故只需对基因1、基因2和基因n+1所对应的变量进行处理即可。
由于一个位点只有12比特宽,要想获得非常高精度的可行解,一个基因所包含的位点数为必需满足212R≥Mr,即R≥(lbMr)/12。一般情况下,当R=1,2,3时,可以获得10-4、10-7、10-10级别的精度。
模型式(1)中的变量既包含实数变量,又包含整数变量,其各值的编码方法如下所述。
(1)实变量编码方法
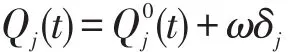
式中,ω为权重;δj称为变量偏差,该值是从下列集合中选择:
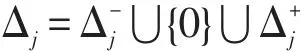
(2)整数变量编码方法
K个0-1整数变量,可以直接采用的编码方法。
2.5 演化算子设计
对图2进行分解,存在下列两种情况:
情况1在时期t,个体从类别A转移到类别C,如图5(a)所示,其中A,C∈{S,I,R},但A≠C。大量的个体类别转移都属于这种情况。例如S→I、I→R、R→S等。

Fig.5 Two classes transfer of an individual at time t图5 时期t个体的两种类别转移情形
为了能使某个体从类别A转移到类别C,将已处于类别C的若干其他个体的某些基因上的位点传给该个体的对应位点,也即使该个体的对应位点具有类别C的个体的基因上的位点。此举实现了该个体从类别A转移到类别C。例如,对于S→I转移,将已处于I类的若干个体的某些基因上的位点传给处于S类的某个体,即可使其感染上传染病,即实现了S→I转移。
情况2在时期t,当个体处于某个类别A时,A∈{S,I,R},并没有发生类别转移,即相当于A→A,如图5(b)所示。图2中的每个节点隐含了图5(b)所示的情形,例如S→S、I→I和R→R。
当某个体处于类别A时,为了能使该个体向好的方向发展,但其类别又保持不变,将已处于同样类别A,但其IPI指数要高于该个体IPI指数的若干个强壮个体的某些基因上的位点传给该个体的对应基因上的位点,也就是将IPI指数高的强壮个体向IPI指数低的虚弱个体传递强壮基因的信息,使得这些虚弱个体能向好的方向发展。
从易感、发病和治愈的个体中分别随机挑选出L个个体,L≥1,u∈{S,I,R},这些个体的IPI指数要高于当前个体i的IPI指数,分别形成强壮易感者集合、染病者集合和治愈者集合CPu={Xj1(t),Xj2(t),…,XjL(t)}。
(1)S-I算子设计。设当前易感个体i被感染,变为染病者,其类别从S→I,操作步骤如下:
(1.1)从W个致病基因中以概率p(p<E0)随机挑选出若干个致病基因,E0表示个体的致病基因被选中的最大概率,即基因被病毒攻击的最大概率。
(1.2)若致病基因j被选中,则:
(1.2.1)从致病基因j的R个位点中随机选出一个位点k与Y进行异或运算,记录Inf(i,1)=j,Inf(i,2)=k。其中,若j≤n,则Y=111111;若n<j≤n+K,则Y=1。此操作表示传染病Z攻击个体i的DNA。此步骤称为基因位点操作。
(1.2.2)从强壮染病者集合CPI中随机挑选出一个个体s,将其对应性状表中的第j列的状态值传给个体i。如图6所示,个体i的第j个致病基因原来的状态值vi转变为个体s的第j个致病基因原来的状态值vs。该步骤相当于染病者传播病毒信息给易感者。此步骤称为个体性状操作。
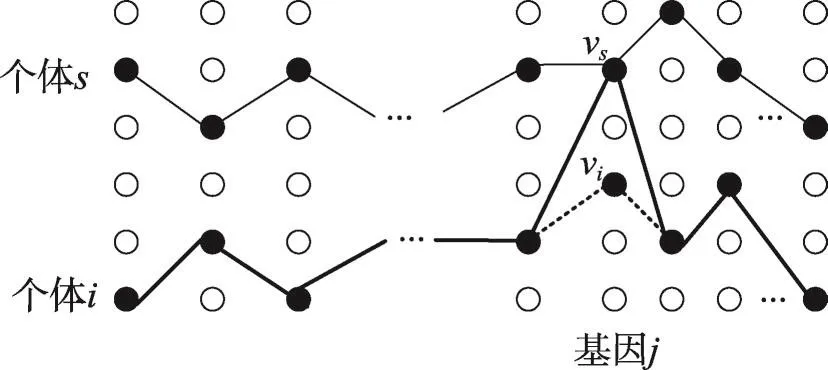
Fig.6 State value vitransferring into vsof the j th disease gene of individual i图6 个体i的第j个致病基因状态值vi变为vs
(2)I-R算子设计。设当前染病个体i被治愈,变为治愈者,其类别从I→R,操作步骤如下:
(2.1)将个体i的所有发生变异的致病基因位点进行复原,即查找个体i的Inf(i,1)和Inf(i,2),找到个体i的发生变异的致病基因和位点,将其与Y再次进行异或运算,即可实现复原。
(2.2)从W个免疫基因中以概率p(p<F0)随机挑选出若干个免疫基因,F0表示个体的免疫基因被选中的最大概率,F0=E0。若免疫基因j被选中,则:
(2.2.1)从免疫基因j的R个位点中随机选出一个位点k与Y进行异或运算,记录Rec(i,1)=j,Rec(i,2)=k。其中,若j≤n,则Y=111111;若n<j≤n+K,则Y=1。此操作表示治愈者获得免疫。
(2.2.2)从强壮染病者集合CPR中随机挑选出一个个体s,将其对应性状表中的第j列的状态值传给个体i。如图6所示。该步骤相当于强壮治愈者将其免疫信息传给染病者,使其治愈并获得免疫。
(3)R-S算子设计。设当前治愈者个体i失去免疫力,变为易感者,其类别从R→S,操作步骤如下:
(3.1)将个体i的所有使其获得免疫力的免疫基因位点进行复原,即查找个体i的Rec(i,1)和Rec(i,2),找到个体i的使其获得免疫力的免疫基因和位点,将这些免疫基因位点与Y再次进行异或运算,即可实现复原,从而使其丧失免疫力。
(3.2)从W个免疫基因中以概率p(p<F0)随机挑选出若干个免疫基因,若免疫基因j被选中,则从强壮易感者集合CPS中随机挑选出一个个体s,将其对应性状表中的第j列的状态值传给个体i。如图6所示。该步骤相当于强壮易感者将其易感信息传给治愈者,使其治愈并丧失免疫力。
(4)S-S算子、I-I算子、R-R算子设计。因这3个算子不存在类别转换,故不进行基因位点操作,但要进行个体性状操作。这3个算子的设计方法类似,其设计方法为:设当前个体i的类别为u,u∈{S,I,R},从个体i的W个基因中以概率p(p<E0)随机挑选出若干个基因,若基因j被选中,则从该类别的强壮者集合CPu中随机挑选出一个个体s,将其对应个体性状表中的第j列的性状值传给个体i。如图6所示。该步骤相当于强壮者将其特有的信息传给当前个体i,使该个体变为更强状态。此操作相当于弱者向强者学习,使自身变强壮。W个基因选择方法是:若u=S,则选W个未染病的致病基因;若u=I,则选W个已染病的致病基因;若u=R,则选W个已免疫的免疫基因。
(5)生长算子设计。个体i的生长算子可以描述为:
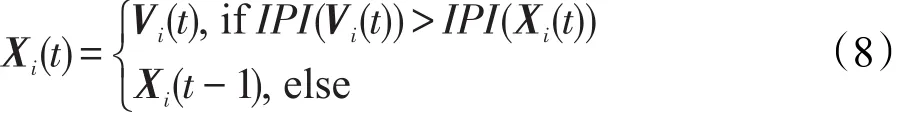
式中,Vi(t)是Xi(t)的一个副本,用于临时保存Xi(t)的值;Xi(t)=(xi1(t),xi2(t),…,xin(t));函数IPI(Vi(t))和IPI(Xi(t))按式(4)计算。
2.6 SIR-DNA算法构造方法
算法1 SIR-DNA算法
(1)初始化:①令时期t=0,最大迭代次数G=108,N=100~200,精度ε=10-8,R=3~5,L=3~ 5,F0=E0=0.000 1~0.01;②初始化N个正常个体X1(0),X2(0),…,XN(0);③在N个正常个体中随机选择30%的个体使其致病;④在剩下的正常个体中随机选择30%的个体使其获得免疫力。
(2)计算ai=Rand(0,1),bi=Rand(0,1),ci=Rand(0,1);Si(0)=ai/(ai+bi+ci),Ii(0)=bi/(ai+bi+ci),Ri(0)=1-Si(0)-Ii(0),i=1,2,…,N。
(3)计算个体i的SIR类别:SIRi(0)=GetSIR(Si(0),Ii(0),Ri(0)),i=1,2,…,N;函数GetSIR()用于确定个体i将处于何种类别。
(4)执行下列操作:


(5)结束。
函数GetSIR(pS,pI,pR)的定义如下:

2.7 SIR-DNA算法的降维特征的理论分析
将式(1)的目标函数展开成如下形式:
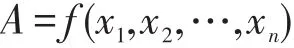
SIR-DNA算法的求解过程最终归结为不断修改变量x1,x2,…,xn的取值,使得目标函数值A不断向理论最优解靠近。在演化过程中,n个变量中的每个变量取值的修改过程是相互独立的;当一个变量的取值被修改后,只能使目标函数值A向3个方向变化:比当前值更好,比当前值更差,与当前值一样。不妨假定这3种情形出现的概率相同,均为1/3。假设SIR-DNA算法每次进化有m个变量的取值同时改变,则能使目标函数值A向好的方向改变的概率为p=1/3m。
显然,当m=n时,即每次进化时n个变量的取值同时改变,此时能使目标函数值A向好的方向发展的概率最低,即p=3-n。因此,每次进化时同时对n个变量的取值进行更新是最不可取的策略,该策略既耗时,又使目标函数值A向好的方向发展的概率达到最低。
另一方面,当m=1时,即每次进化时只修改1个变量的取值,此时能使目标函数值A向好的方向发展的概率最大,即p=1/3。因此,每次进化时只对1个变量的取值进行更新是可取的策略,该策略能使A向好的方向发展的概率达到最大。
上面的分析给出了SIR-DNA算法在求解过程中每次处理变量数的一般规律,这是一个普适规律。然而,由于求解过程的复杂性,每次只修改1个变量的取值不一定性能最佳,但一般规律是:无论n多大,每次参与运算的变量数都不要超过10个,即1≤nE0<10。
上述分析表明,SIR-DNA算法具有天然降维特征。
2.8 SIR-DNA算法时间复杂度
SIR-DNA算法的时间复杂度如表3所示。

Table 3 Time complexity of SIR-DNA表3 SIR-DNA算法的时间复杂度计算表
2.9 SIR-DNA算法收敛性和稳定性分析
从S-S、S-I、I-I、I-R、R-R、R-S算子的定义可知,任何一个试探解的新一代的生成只与该试探解的当前状态有关,而与该试探解以前是如何演变到当前状态的历程无关,因而SIR-DNA算法的演化过程具有Markov特性。因老的试探解无需保留,故可以使算法的空间复杂度降到最低。
从生长算子的定义可知,若从当前位置出发,下一步搜索方向只有两个,即要么向比当前位置更好的方向搜索,要么留在当前位置不动。因此,SIRDNA算法具有“步步不差”的搜索特征。
由于SIR-DNA算法的演化过程具有Markov特性和“步步不差”特性,根据文献[13]可得如下定理。
定理1 SIR-DNA算法具有全局收敛性。
SIR-DNA算法的稳定性依赖于SIR传染病动力学模型的解的稳定性,即微分方程组(2)的解的稳定性。文献[12]给出了该模型的解保持全局稳定性的条件,本文不再赘述。于是,SIR传染病模型可以帮助SIR-DNA算法选择最合理的参数实现稳定收敛。
3 SIR-DNA算法与其他算法比较
本文使用CEC2013[14]所提供的国际上通用的基准函数来测试SIR-DNA算法的性能,本文选择的6个基准函数,如表4所示。在表4中,O是一个n维决策向量。这里用SIR-DNA算法去求解表4所示的基准函数,其参数是n=50,N=200,O的值随机产生。选择7种优化算法与SIR-DNA算法进行比较,这些算法如表5所示。
用这些算法独立求解每个基准函数51次,计算结果如表6所示。表6的排名1是按平均最佳目标函数值排序,排名2是按平均最佳目标函数值和CPU时间排序。表7是8种算法所得最优解的非参数Wilcoxon秩和检验。表中,h-val=1表明SIR-DNA算法能够以99%的概率优于其他算法,h-val=-1表示SIR-DNA算法明显劣于其他算法,而h-val=0表示SIR-DNA算法与其他算法的结果差异不显著。
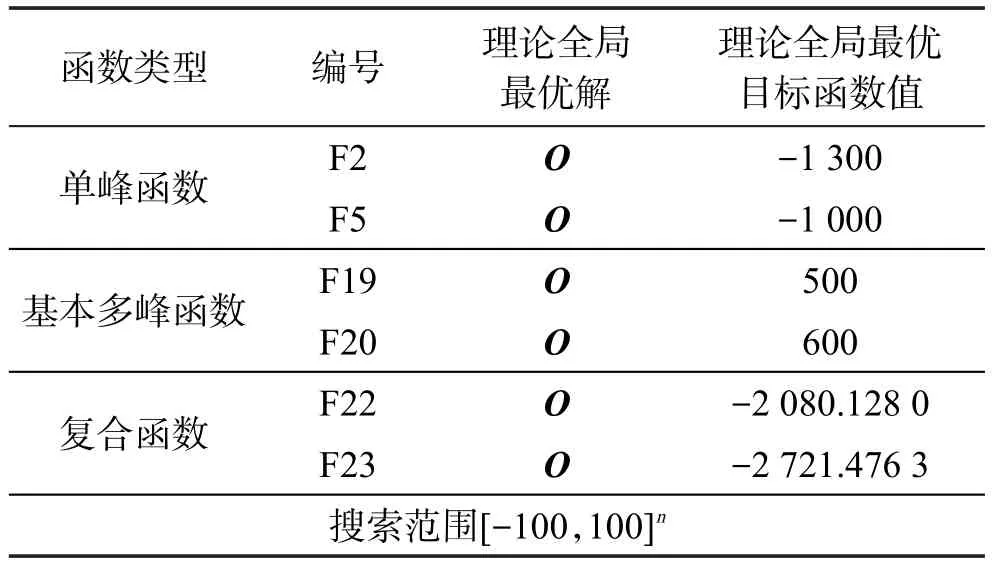
Table 4 Benchmark functions表4 基准函数
从表6中可以看出,这8个算法按精度排序如下:
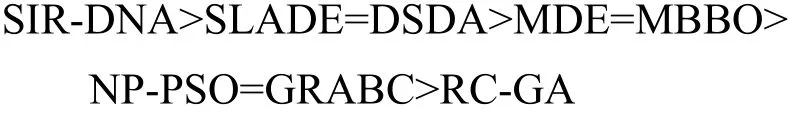
按精度和CPU耗时的排序如下:
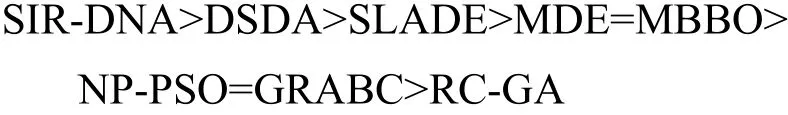
从表7中可以知道,SIR-DNA算法的性能明显优于7种被比较的算法。
图7(a)~(c)说明8个算法求解F2、F20和F22时的样本收敛曲线,其中的水平和垂直轴采用对数刻度。从表6中可以看出,当SIR-DNA算法求解F2、F20时,其发现最优解所需时间要比被比较算法少;当SIR-DNA算法求解F22时,其发现最优解的精度和所需时间稍逊于DSDA算法,但比其他算法要好。综合看来,SIR-DNA算法的综合性能要优于被比较算法,表明其计算速度快。从图7(a)~(b)可以看出,SIR-DNA算法的收敛曲线大部分在被比较算法的左侧,表明其收敛速度很快;从图7(c)可以看出,SIRDNA算法的收敛曲线在被比较算法的右侧,表明其收敛速度慢于某些算法,但是只有SIR-DNA算法获得的最优解最好。

Table 5 Parameters of 7 compared algorithms表5 7种参与比较的算法的参数
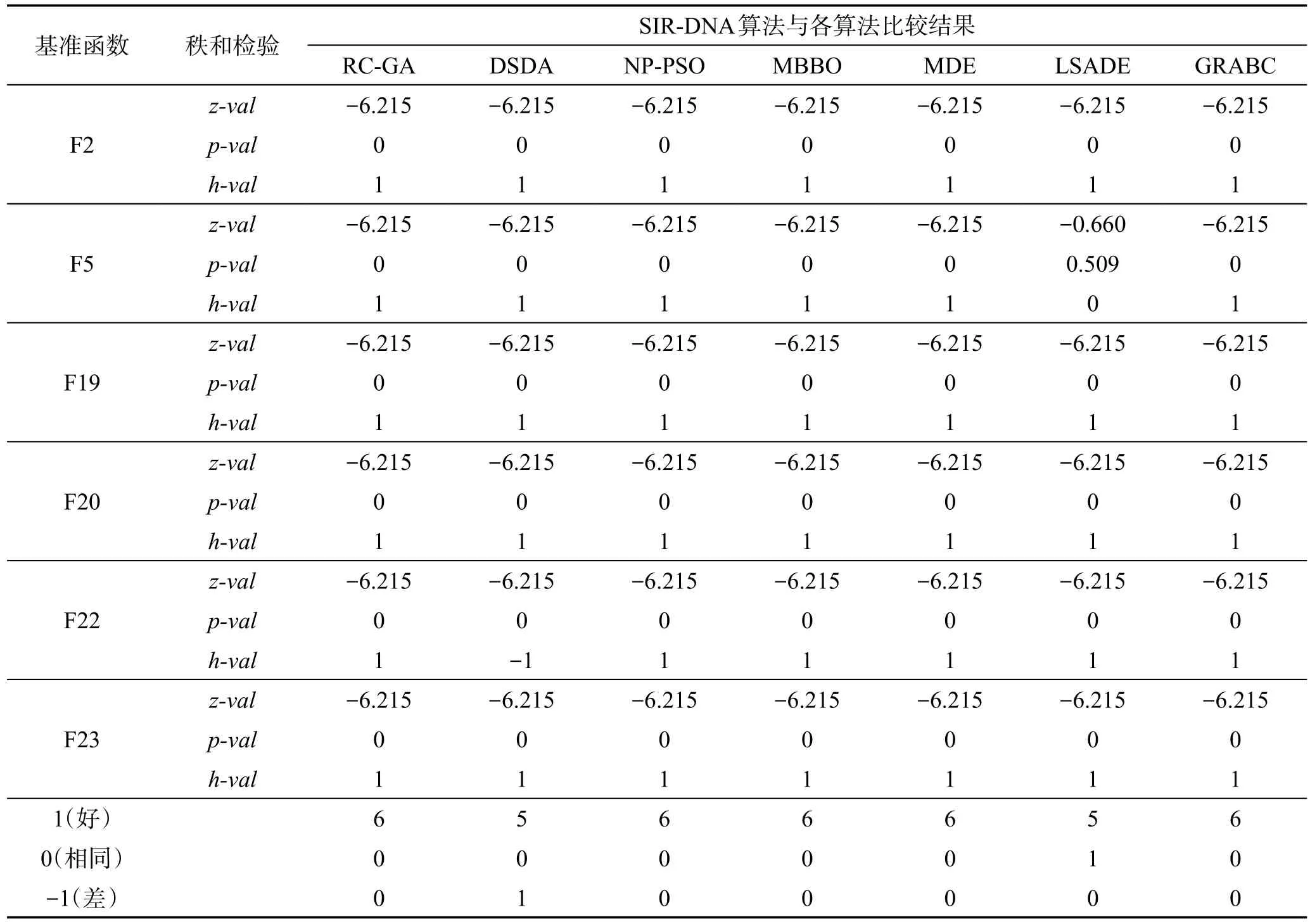
Table 7 Results comparison of Wilcoxon rank sum test(α=0.01)表7 Wilcoxon秩和检验结果比较(α=0.01)
4 结论
本文采用SIR传染病动力学理论与DNA分子结构理论相结合的方法构建出SIR-DNA算法。在该算法中,SIR传染病模型的优势和特性如下:
(1)S-S、S-I、I-I、I-R、R-R、R-S算子不与优化问题相关,从而使SIR-DNA算法具有通用性。
(2)利用传染病传播时所诱发的6个状态转移,从多种角度天然地实现个体之间信息的充分交换,降低了个体陷入局部最优的概率。
(3)因病毒每次攻击的是个体的很少部分特征,每次运算只涉及到很少一部分特征参与运算,从而实现天然降维。
(4)SIR-DNA算法利用SIR传染病动力学理论,使搜索过程达到生态稳定时出现收敛,而SIR传染病模型可以帮助算法选择合理的参数实现收敛。
在SIR-DNA算法中,由于采用SIR传染病模型作为基础,从一个新的角度构造了能反映DNA特征的优化算法,其特征在于:
(1)SIR-DNA算法不是直接与DNA算法进行结合,从而避免了DNA算法的缺陷,即DNA计算时需要借助生物实验,无法求解高维优化问题等。
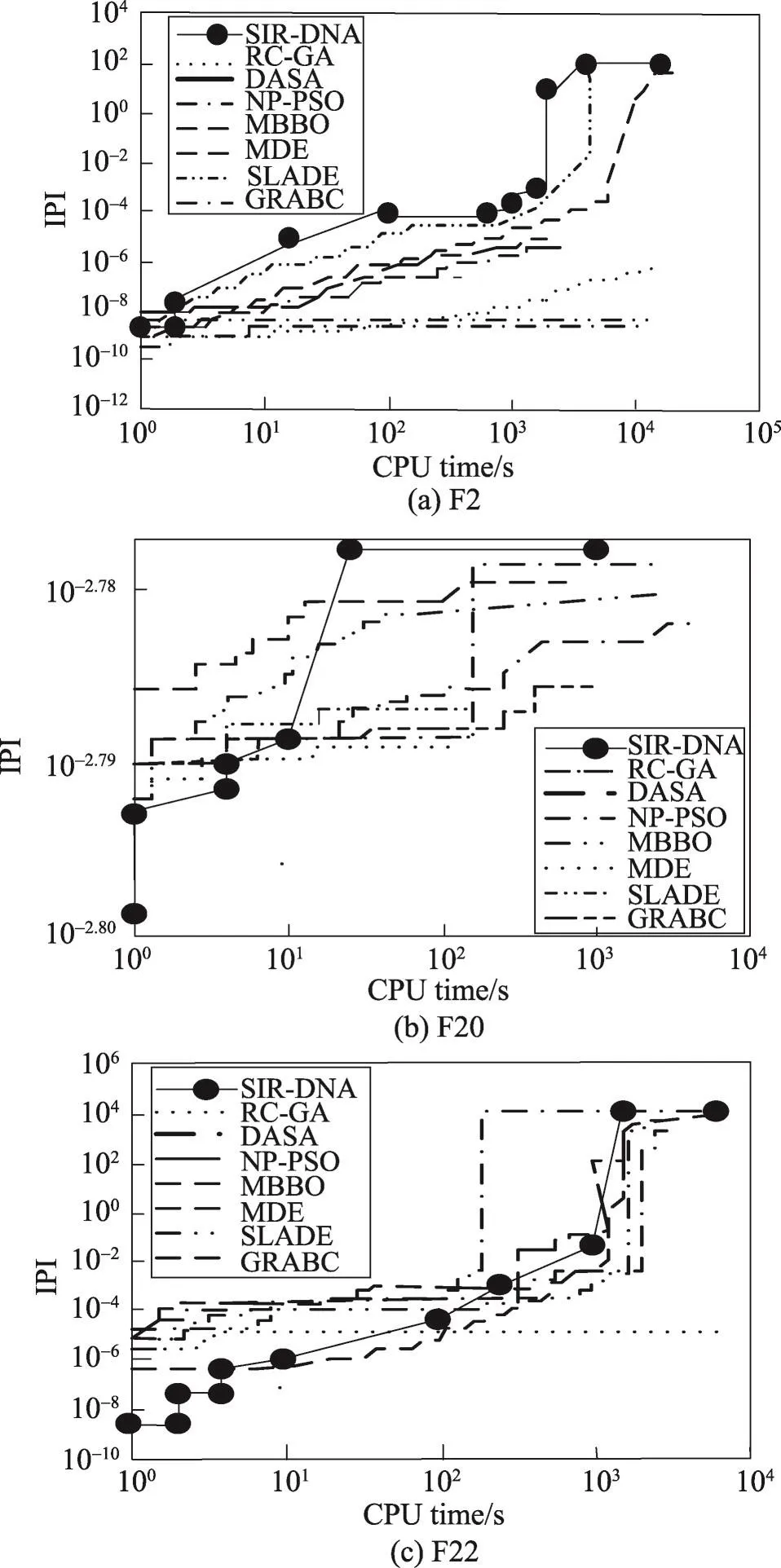
Fig.7 Convergence curves when 8 algorithms solve F2、F20、F22图7 8个算法求解F2、F20、F22时的收敛曲线
(2)SIR-DNA算法纳入DNA特征的方法是,将DNA中的基因和位点进行编码,采用随机修改关键基因及其位点的方法来模拟病毒对DNA的攻击或获得免疫力,从而无需生物实验即可进行计算。
迄今为止,已经发现了数以千计的传染病。本文的SIR-DNA算法为将这些传染病转化为能求解的非常复杂的优化问题的群智能优化算法提供了参考。
[1]Adleman L M.Molecular computation of solution to combinatorial problems[J].Science,1994,266(5187):1021-1024.
[2]Zhang Xuncai,Zhao Hailan,Cui Guangzhao,et al.Research advances and prospect of DNAcomputing[J].Computer Engineering andApplications,2007,43(10):44-47.
[3]Takahashi K,Yaegashi S,Asanuma H,et al.Photo-and thermoregulation of DNA nanomachines[C]//LNCS 3892:Proceedings of the 11th International Workshop on DNA Computing,London,Jun 6-9,2005.Berlin,Heidelberg:Springer,2005:336-346.
[4]Wu Fan,Li Kenli.An algorithm in tile assembly model forNqueen problem[J].Acta Electronica Sinica,2013,41(11):2174-2180.
[5]Li Kenli,Luo Xing,Wu Fan,et al.An algorithm in tile assembly model for maximum clique problem[J].Journal of Computer Research and Development,2013,50(3):666-675.
[6]Yao Qing'an,Zheng Hong,Wang Hongmei.DNA computing model for shortest path problem based onk-armed molecule and sticker operation[J].Journal of Jilin University:Information Science Edition,2014,32(6):653-656.
[7]Xu Jinglei,Zhao Hongchao,Liu Xiyu.Closed circle DNA algorithm of traveling salesman problem[J].Computer Engineering&Science,2014,36(1):111-114.
[8]Ma Ying,Yin Zhixiang.DNA computing model for the graph vertex coloring problem by plasmids[J].Journal of Anhui University of Science and Technology:Natural Science,2015,35(2):64-67.
[9]Wu Xue,Song Chenyang,Zhang Nan,et al.DNA algorithm for maximum matching problem based on sticker computation model[J].Computer Science,2013,40(12):127-132.
[10]Kermack W O,Mckendrick A G.Contributions to the mathematical theory of epidemics[J].Proceedings of the Royal Society of London:SeriesA,1927,115(772):700-721.
[11]Kermack W O,Mckendrick A G.Contributions to the mathematical theory of epidemics(II):the problem of endemicity[J].Proceedings of the Royal Society of London,1932,138(795):55-83.
[12]Wang Qian,Wang Tingting.Stability analysis of a SIRS epidemic model[J].Journal of Capital Normal University:Natural Science Edition,2016,37(2):5-11.
[13]Huang Guangqiu.SIS epidemic model-based optimization[J].Journal of Computation Science,2014,5(1):32-50.
[14]Liang J J,Qu B Y,Suganthan P N,et al.Problem definitions and evaluation criteria for the CEC 2013 special session on real-parameter optimization,201212[R].Singapore:Nanyang Technological University,2013.
[15]Chuang Yaochen,Chen C T,Hwang C.A simple and efficient real-coded genetic algorithm for constrained optimization[J].Applied Soft Computing,2016,38:87-105.
[16]Korošec P,Šilc J,Filipič B.The differential ant-stigmergy algorithm[J].Information Sciences,2012,192:82-97.
[17]Beheshti Z,Shamsuddin S M.Non-parametric particle swarm optimization for global optimization[J].Applied Soft Computing,2015,28:345-359.
[18]Al-Roomi A R,El-Hawary M E.Metropolis biogeographybased optimization[J].Information Sciences,2016,360:73-95.[19]Mukherjee R,Debchoudhury S,Das S.Modified differential evolution with locality induced genetic operators for dynamic optimization[J].European Journal of Operational Research,2016,253(2):337-355.
[20]Zhao Zhiwei,Yang Jingming,Hu Ziyu,et al.A differential evolution algorithm with self-adaptive strategy and control parameters based on symmetric Latin hypercube design for unconstrained optimization problems[J].European Journal of Operational Research,2016,250(1):30-45.
[21]Li Genghui,Cui Laizhong,Fu Xianghua,et al.Artificial bee colony algorithm with gene recombination for numerical function optimization[J].Applied Soft Computing,2017,52:146-159.
附中文参考文献:
[2]张勋才,赵海兰,崔光照.DNA计算的研究进展及展望[J].计算机工程与应用,2007,43(10):44-47.
[4]吴帆,李肯立.基于自组装的N皇后问题DNA计算算法[J].电子学报,2013,41(11):2174-2180.
[5]李肯立,罗兴,吴帆,等.基于自组装模型的最大团问题DNA计算算法[J].计算机研究与发展,2013,50(3):666-675.
[6]姚庆安,郑虹,王红梅.基于k-臂分子求解最短路径的DNA计算模型[J].吉林大学学报:信息科学版,2014,32(6):653-656.
[7]徐京雷,赵洪超,刘希玉.旅行商问题的闭环DNA算法[J].计算机工程与科学,2014,36(1):111-114.
[8]马莹,殷志祥.图顶点着色问题的质粒DNA计算[J].安徽理工大学学报:自然科学版,2015,35(2):64-67.
[9]吴雪,宋晨阳,张楠,等.最大匹配问题的粘贴DNA算法[J].计算机科学,2013,40(12):127-132.
[12]王茜,王婷婷.SIRS传染病模型的稳定性分析[J].首都师范大学学报:自然科学版,2016,37(2):5-11.
猜你喜欢
分子催化(2022年1期)2022-11-02
青年文学家(2022年13期)2022-07-06
陶瓷学报(2021年4期)2021-10-14
昆明医科大学学报(2021年8期)2021-08-13
烟草科技(2021年6期)2021-06-24
少儿画王(3-6岁)(2020年4期)2020-09-13
青海湖(2020年6期)2020-09-07
电脑知识与技术(2018年19期)2018-11-01
诗选刊(2016年9期)2016-11-26
微型计算机(2009年4期)2009-12-23
