
二芳甲基取代的大位阻酚的制备及其金属化研究
2018-04-21 08:16罗晓军祝飞凤范国康
西南民族大学学报(自然科学版) 2018年2期
罗晓军,祝飞凤,范国康,雷 浩
(暨南大学化学与材料学院,广东 广州 510632)
大空间位阻的单齿配体(如烷氧基、酚基、胺基、芳基、烷基、卡宾等)已被证明在合成稳定的低配位数过渡金属配合物中具有重要作用[1].烷氧基或酚基配体的氧原子既是σ电子给体,又可做π电子给体,通常可用于稳定缺电子的前过渡金属中心.当这类配体只以σ给电子作用与金属中心配位时,氧原子上的孤对电子可能导致金属中心之间的寡聚反应[2-6],最终使得配体从金属中心解离[7-9].通过在配体结构中引入大空间位阻的取代基团,例如在苯酚的邻位引入异丙基、苯基、叔丁基等[10-13],可以极大地提高配合物金属中心的稳定性,抑制配合物分子间的寡聚反应,为低配位数金属配合物的制备和分离提供更多的可能性.大位阻酚基配体正是由于其自身电子性质和空间位阻的高度可调性而受到众多科研工作者的重视,常被用作合成低配位金属配合物的支撑配体[14-17].
Kawaguchi等人在2011年通过Fe{N(SiMe3)2}2与两倍量的大位阻酚HOArAdR(Ad为金刚烷基,R为甲基或异丙基)的反应制备了二配位亚铁化合物Fe(OArAdR)2.该酚基配体中具有较大刚性和空间位阻的金刚烷基是成功合成二配位亚铁化合物的关键.该亚铁配合物能与AdN3反应,生成含有Fe=N键的高价态配合物.产物中的Fe(IV)中心不稳定,能进一步活化酚基配体上金刚烷基的C-H键,生成新的C-N键和N-H键[18].
Mindiola等人报道合成了一种新型大位阻酚配体.这种配体可以通过二苯甲醇和苯酚衍生物在无溶剂条件下反应得到,合成方法简单,产率和产品纯度都很高[19].在本文中,我们根据前人的思路进一步设计并合成了一系列新型的二芳甲基取代的大位阻酚化合物,并通过其与正丁基锂的脱质子反应进一步合成了对应的酚基锂配合物.
1 实验部分
1.1 仪器及药品
主要仪器有惰性气氛手套箱,型号Super(1220/750),米开罗那(中国)有限公司;溶剂纯化系统,PureSolv MD5,美国 Innovative Technology;核磁共振波谱仪,300 MHz,布鲁克.
所用药品4-甲基苯酚、对溴甲苯、二苯甲醇、4-叔丁基溴苯、正丁基锂(己烷溶液)、甲酸乙酯、氯化锌等均购自萨恩化学技术(上海)有限公司;镁条、氯化钠、氯化铵、无水碳酸钾、无水硫酸镁等均购自天津市大茂化学试剂厂.甲酸乙酯在使用前以CaH2干燥后常压蒸馏提纯,4-叔丁基溴苯使用前经过减压蒸馏提纯,无水无氧操作中用到的有机溶剂均经过创新科技PureSolv MD5溶剂纯化系统进行干燥纯化后使用.
1.2 实验方法
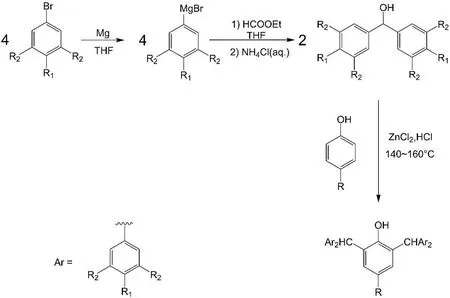
路线图1 化合物1-8的合成路线Scheme 1 Synthetic route of compounds 1-8
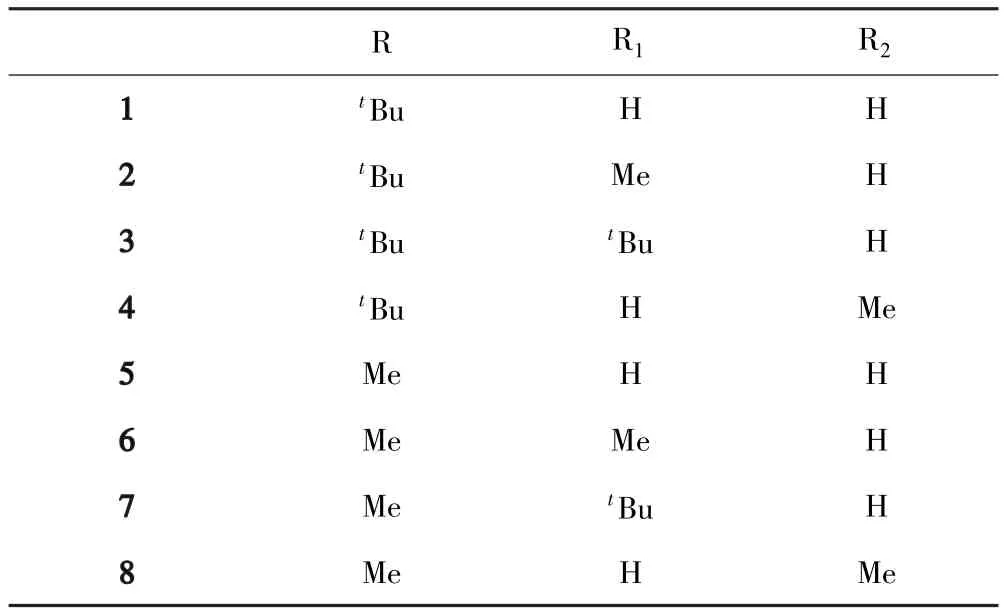
表1 化合物1-8的取代基组成Table 1 Compositions of compounds 1-8
1:在500 mL圆底烧瓶中加入13.51 g(73.3 mmol)二苯甲醇和5.52 g(36.6 mmol)对叔丁基苯酚,加热至140℃,逐滴加入预先混合好的催化剂(2.58 g ZnCl2和3.14 mL浓盐酸的混合物),维持温度在140~160℃反应2小时.用二氯甲烷萃取,分别用蒸馏水和饱和食盐水洗涤两次,以无水碳酸钾干燥.抽滤,蒸干溶剂,向所得黄色固体中加入约100 mL甲醇回流2小时.冷却后抽滤,再用少量甲醇洗涤,得到白色固体产品.产量13.29 g,产率75.2%.1H NMR(300 MHz,CDCl3,TMS):δ 7.31-7.26(m,8H,苯环 H),7.24-7.19(m,4H,苯环 H),7.10(d,8H,苯环H),6.71(s,2H,苯环 H),5.67(s,2H,-CHPh2),4.38(s,1H,-OH),1.03(s,9H,-C(CH3)3).
2:合成分为两步.
第一步:称量4.68 g(192.6 mmol)镁条加入到500 mL三口烧瓶中,装上回流冷凝管,在氮气保护下加入约45 mL THF后,加入一小粒碘,搅拌加热,直至溶液颜色不再变化为止.再逐滴加入对甲基溴苯(24.32 g,142.2 mmol)的THF溶液,溶液由无色变灰色,最后为黑色,加热回流12小时.冷却至室温,逐滴加入甲酸乙酯(5.31 g,71.7 mmol),回流半个小时.冷却至室温后再加入约200 mL NH4Cl水溶液,以CH2Cl2萃取(3×100 mL),用无水 MgSO4干燥.抽滤,蒸干溶剂,得到浅黄色固体,在己烷中重结晶,得到白色固体二(4-甲基苯基)甲醇.产量10.80 g,产率66%.1H NMR(300 MHz,CDCl3,TMS):δ 7.27-7.25(m,4H,苯环 H),7.14(d,4H,苯环 H),5.79(d,1H,-CHPh2),2.33(s,6H,-CH3),2.11(d,1H,-OH).
第二步:与化合物1的合成步骤类似,加入的药品和具体用量分别为10.80 g(50.9 mmol)二(4-甲基苯基)甲醇和3.82 g(25.5 mmol)对叔丁基苯酚、催化剂(1.76 g ZnCl2和3 mL浓盐酸的混合物),最终得到浅黄色固体产品.产量10.20 g,产率74%.1H NMR(300 MHz,CDCl3,TMS):δ 7.07(d,8H,苯环 H),6.98(d,8H,苯环 H),6.72(s,2H,苯环 H),5.58(s,2H,-CHPh2),4.46(s,1H,-OH),2.32(s,12H,-CH3),1.04(s,9H,-CH3).
3:合成分为两步.
第一步:与化合物2的第一步合成步骤类似,加入的药品和具体用量分别为3.65 g(150 mmol)镁条、对叔丁基溴苯25 g(117.3 mmol)、甲酸乙酯3.90 g(52.6 mmol),得到白色固体二(4-叔丁基苯基)甲醇.产量 11.68 g,产率 75%.1H NMR(300 MHz,CDCl3,TMS):δ 7.38-7.30(m,8H,苯环 H),5.81(d,1H,-CHPh2), 2.14(d, 1H,-OH), 1.30(s, 18H,-C(CH3)3).
第二步:与化合物1的合成步骤类似,加入的药品和具体用量分别为10.86 g(35.7 mmol)二(4-叔丁基苯基)甲醇和2.69 g(17.9 mmol)对叔丁基苯酚、催化剂(1.22 g ZnCl2和2 mL浓盐酸的混合物),得到浅黄色固体产品.产量9.86 g,产率77.8%.1H NMR(300 MHz,CDCl3,TMS):δ 7.29(d,8H,苯环 H),7.04(d,8H,苯环 H),6.70(s,2H,苯环 H),5.62(s,2H,-CHPh2), 4.47(s, 1H,-OH), 1.30(s, 36H,-C(CH3)3),1.04(s,9H,-C(CH3)3).
4:合成分为两步.
第一步:与化合物2的第一步合成步骤类似,加入的药品和具体用量分别为3.97 g(163.2 mmol)镁条、3,5-二甲基溴苯 24.52 g(132.5 mmol)、甲酸乙酯4.40 g(59.4 mmol),得到白色固体二(3,5-二甲基苯基)甲醇.产量9.71 g,产率73%.1H NMR(300 MHz,CDCl3,TMS):δ 7.00(s,4H,苯环 H),6.90(s,2H,苯环 H),5.70(d,1H,-CHPh2),2.30(s,12H,-CH3),2.08(d,1H,-OH).
第二步:与化合物1的合成步骤类似,加入的药品和具体用量分别为9.71 g(40.4 mmol)二(3,5-二甲基苯基)甲醇和3.03 g(20.2 mmol)对叔丁基苯酚、催化剂(1.38 g ZnCl2和3 mL浓盐酸的混合物),得到浅黄色固体产品.产量9.86 g,产率78%.1H NMR(300 MHz,CDCl3,TMS):δ 6.85(s,4H,苯环 H),6.77(s,2H,苯环 H),6.72(s,8H,苯环 H),5.51(s,2H,-CHPh2),4.54(s,1H,-OH),2.24(s,24H,-CH3),1.09(s,9H,-C(CH3)3).
5:与化合物1的合成步骤类似,加入的药品和具体用量分别为27.02 g(146.5 mmol)二苯甲醇和7.92 g(73.2 mmol)对甲基苯酚、催化剂(5.04 g ZnCl2和6.3 mL浓盐酸的混合物),得到白色固体产品.产量28.85 g, 产率 94.5%.1H NMR(300 MHz, CDCl3,TMS):δ 7.31-7.26(m,8H,苯环 H),7.24-7.19(m,4H,苯环 H),7.10(d,8H,苯环 H),6.49(s,2H,苯环H),5.67(s,2H,-CHPh2),4.37(s,1H,-OH),2.07(s,3H,-CH3).
6:与化合物1的合成步骤类似,加入的药品和具体用量分别为5.98 g(29.8 mmol)二(4-甲基苯基)甲醇和1.62 g(15.0 mmol)对甲基苯酚、催化剂(1.02 g ZnCl2和1.5 mL浓盐酸的混合物),得到暗黄色固体产品.产量 5.53 g,产率 78%.1H NMR(300 MHz,CDCl3,TMS):δ 7.08(d,8H,苯环 H),6.98(d,8H,苯环 H),6.49(s,2H,苯环 H),5.58(s,2H,-CHPh2),4.44(s,1H,-OH),2.32(s,12H,-CH3),2.08(s,1H,-CH3).
7:与化合物1的合成步骤类似,加入的药品和具体用量分别为7.06 g(23.8 mmol)二(4-叔丁基苯基)甲醇和1.29 g(11.9 mmol)对甲基苯酚、催化剂(1.29 g ZnCl2和1.5 mL浓盐酸的混合物),得到暗黄色固体产品.产量 6.87 g,产率 90%.1H NMR(300 MHz,CDCl3,TMS):δ 7.27(t,8H,苯环 H),7.02(d,8H,苯环 H),6.54(s,2H,苯环 H),5.61(s,2H,-CHPh2),4.45(s,1H,-OH),2.10(s,3H,-CH3),1.30(s,36H,-C(CH3)3).
8:与化合物1的合成步骤类似,加入的药品和具体用量分别为3.90 g(16.2 mmol)二(3,5-二甲基苯基)甲醇和0.90 g(8.3 mmol)对甲基苯酚、催化剂(0.57 g ZnCl2和1.5 mL浓盐酸的混合物),得到暗黄色固体产品.产量2.02 g,产率78%.1H NMR(300 MHz,CDCl3,TMS):δ 6.84(s,4H,苯环 H),6.71(s,8H,苯环 H),6.50(s,2H,苯环 H),5.50(s,2H,-CHPh2),4.49(s,1H,-OH),2.24(s,24H,-CH3),2.11(s,3H,-CH3).
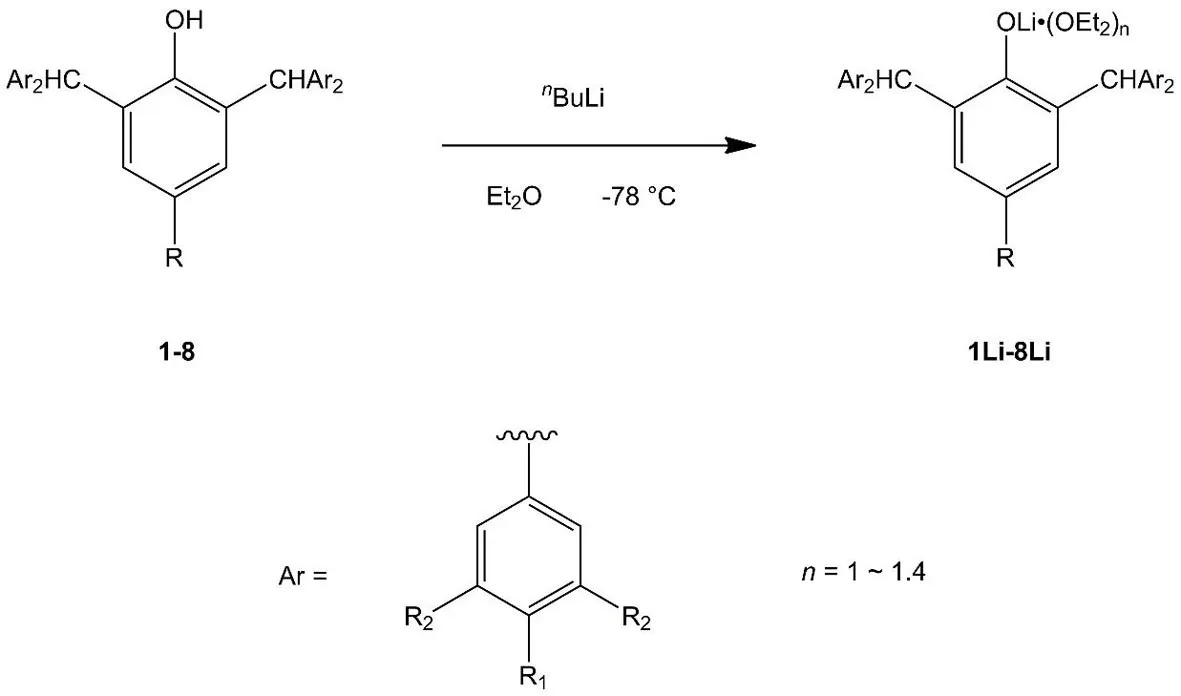
路线图2 化合物1Li-8Li的合成路线Scheme 2 Synthetic route of compounds 1Li-8Li
化合物1Li-8Li的合成需要利用Schlenk技术在N2气氛下完成.
1Li:将化合物1(9.66 g,20 mmol)加入500 mL Schlenk瓶中,在Schlenk线上真空干燥(约70℃)2小时.加入200 mL乙醚将其溶解,在-78℃缓慢加入2.5 M正丁基锂的己烷溶液(9 mL,22.5 mmol),恢复至室温,搅拌12 h.将反应液浓缩至20 mL,放入4℃冰箱中静置,析出的固体用冷的正己烷洗涤2次(10 mL×2),抽干溶剂,得到红色固体产品.产量4.28 g,产率44%.1H NMR(300 MHz,C6D6):δ 7.20(d,8H,苯环 H),7.08(t,8H,苯环 H),6.96(t,6H,苯环 H),5.68(s,2H,-CHPh2),1.18(s,9H,-CH3).
2Li:与化合物1Li的合成步骤类似,加入的药品和具体用量分别为2(4.71 g,8.74 mmol)、2.5 M 正丁基锂的己烷溶液(4.5 mL,11.0 mmol),得到橙红色固体产品.产量 3.73 g,产率 78%.1H NMR(300 MHz,C6D6):δ 7.23(d,8H,苯环 H),7.09(s,2H,苯环 H),6.99(d,8H,苯环 H),5.72(s,2H,-CHPh2),2.07(s,12H,-CH3),1.21(s,9H,-C(CH3)3).
3Li:与化合物1Li的合成步骤类似,加入的药品和具体用量分别为3(7.07 g,10 mmol)、2.5 M正丁基锂的己烷溶液(4.7 mL,11.75 mmol),得到深黄色固体产品.产量 6.61 g,产率 93%.1H NMR(300 MHz,C6D6):δ 7.26-7.19(m,18H,苯环 H),5.89(s,2H,-CHPh2),1.27(s,9H,-C(CH3)3),1.20(s,36H,-C(CH3)3).
4Li:与化合物1Li的合成步骤类似,加入的药品和具体用量分别为4(4.65 g,7.82 mmol)、2.5 M 正丁基锂的己烷溶液(4 mL,10 mmol),得到黄色固体产品.产量 4.0 g,产率 91%.1H NMR(300 MHz,C6D6):δ 7.16(d,2H,苯环H),7.09(s,8H,苯环H),6.70(s,4H,苯环 H),5.69(s,2H,-CHPh2),2.10(s,24H,-CH3),1.23(s,9H,-CH3).
5Li:与化合物1Li的合成步骤类似,加入的药品和具体用量分别为5(3.37 g,8 mmol)、2.5 M正丁基锂的己烷溶液(3.5 mL,8.75 mmol),得到橘红色固体产品.产量 3.01 g,产率 88%.1H NMR(300 MHz,C6D6):δ 7.14(d,8H,苯环 H),7.05(m,8H,苯环 H),6.95(m,4H,苯环 H),6.78(s,2H,苯环 H),5.76(s,2H,-CHPh2),2.04(s,3H,-CH3).
6Li:与化合物1Li的合成步骤类似,加入的药品和具体用量分别为6(1.13 g,2.39 mmol)、2.5 M 正丁基锂的己烷溶液(1.5 mL,3.75 mmol),得到红褐色固体产品.产量 0.91 g,产率 79%.1H NMR(300 MHz,C6D6):δ 7.19(d,8H,苯环 H),6.97(d,8H,苯环 H),6.88(s,2H,-CHPh2),2.09(s,15H,-CH3).
7Li:与化合物1Li的合成步骤类似,加入的药品和具体用量分别为7(4.48 g,7 mmol)、2.5 M正丁基锂的己烷溶液(3.0 mL,7.5 mmol),得到浅黄色固体产品.产量 3.90 g,产率 77%.1H NMR(300 MHz,C6D6):δ 7.19-7.25(m,16H,苯环 H),7.03(s,2H,苯环H),6.00(s,2H,-CHPh2),2.05(s,3H,-CH3),1.21(s,36H,-C(CH3)3).
8Li:与化合物1Li的合成步骤类似,加入的药品和具体用量分别为8(1.6 g,2.89 mmol)、2.5 M正丁基锂的己烷溶液(1.7 mL,4.25 mmol),得到浅黄色固体产品.产量1.62 g,产率69%.1H NMR(300 MHz,C6D6):δ 7.12(s,8H,苯环 H),6.98(s,2H,苯环 H),6.69(s,4H,苯环 H),5.82(s,2H,-CHPh2),2.13(s,3H,-CH3),2.08(d,24H,-CH3).
2 结果与讨论
2.1 化合物1-8的1H NMR谱图分析
本系列化合物主要表征手段为1H NMR,以化合物8为例,如图1所示,在δ 4.41~4.54 ppm之间出现的单峰为酚羟基上的氢质子的吸收峰(A);在δ 2.07~2.13 ppm左右的单峰为主芳环上4-位甲基氢质子的吸收峰(B);在δ 5.50~5.68 ppm之间出现的单峰为二芳甲基上的氢质子的吸收峰(C);在δ 2.16~2.32 ppm之间出现的单峰为二芳甲基所连两个芳环上的甲基氢质子的吸收峰(D);在δ 6.45~6.60 ppm之间出现的单峰为主苯环上两个氢质子的吸收峰(E);在δ 6.64~6.90 ppm之间出现的吸收峰为二芳甲基上的芳环氢质子的吸收峰(F、G).化合物的核磁氢谱结构明晰,各共振吸收峰的化学位移以及峰积分的比例符合预期,说明合成的化合物1-8纯度较高,可用于后续的金属化反应.此外,该系列大位阻酚的分子结构还可以通过单晶X-射线衍射手段进行表征.Mindiola等人发现由于分子间氢键的作用,化合物1采取二聚体的固态分子结构[19].
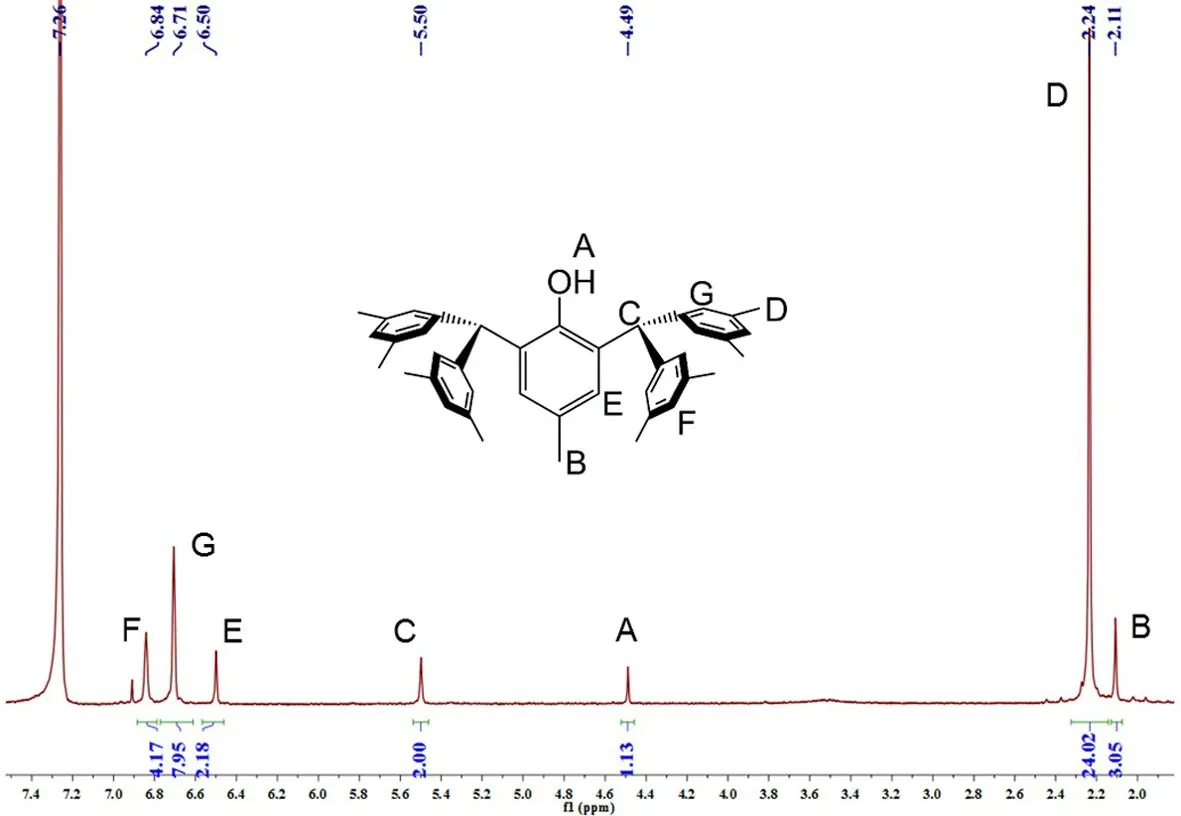
图1 化合物8的1H NMR谱图Fig.1 1H NMR spectrum of compound 8
2.2 化合物1Li-8Li的1H NMR谱图分析
本系列酚基锂化合物主要通过1H NMR手段进行表征,以化合物8Li为例,如图2所示.与化合物8的1H NMR谱图相比,8Li的核磁氢谱中不存在酚羟基的共振峰,这说明脱质子反应进行得很完全.具体来说,在δ 2.10~2.13 ppm之间的吸收峰为芳环上取代烷基氢质子吸收峰(A);在δ 5.68~6.00 ppm之间宽而低矮的单峰为二芳甲基上的氢质子吸收峰(B);在δ 2.05~2.10 ppm之间出现的单峰为二芳甲基所连两个芳环上的甲基氢质子的吸收峰(C);在δ 6.97~6.99 ppm之间出现的单峰为主苯环上两个氢质子的吸收峰(D);在δ 6.65~6.75 ppm和δ 7.10~7.15 ppm之间出现的吸收峰为二芳甲基上的芳环氢质子的吸收峰(E、F).此外,δ 0.93~1.14 ppm 和 δ 3.06~3.29 ppm处均同时出现共振吸收峰,氢原子个数比例约为2:3,说明产物中含有与锂原子配位的溶剂乙醚分子.根据核磁谱图积分结果计算,化合物1Li~8Li中酚基锂配合物与乙醚的摩尔比约为1:1~1:1.4.这些乙醚分子不能通过真空干燥的方式除去,说明其与锂金属中心具有较强的配位作用.Mindiola等人曾报道了类似的酚基钠化合物1Na的晶体结构[19].

图2 化合物8Li的1H NMR谱图Fig.2 1H NMR spectrum of compound 8Li
3 结论
通过4-叔丁基苯酚或4-甲基苯酚与二苯甲醇及其衍生物的反应合成了一系列二芳甲基取代的大位阻酚化合物,并采用核磁共振氢谱进行了表征,证实均得到了较纯的产物.通过酚化合物与正丁基锂的反应制备了相应的酚基锂化合物,核磁共振氢谱确认了其组成和结构,为进一步制备含有大位阻酚基的低配位数过渡金属配合物提供了物质基础.
[1]POWER P P.Some highlights from the development and use of bulky monodentate ligands[J].Journal of Organometallic Chemistry,2004,689(24):3904-3919.
[2]NIELSON A J,SHEN C,WATERS J M.Molecular engineering of coordination pockets in chloro-tris-phenoxo complexes of Titanium(IV)[J].Polyhedron,2006,25(10):2039-2054.
[3]GOWDA R R,CHAKRABORTY D,RAMKUMAR V.Aryloxy and benzyloxy compounds of zirconium:synthesis,structural characterization and studies on solvent-free ring-opening polymerization of-caprolactone and δ-valerolactone[J].Journal of Organometallic Chemistry,2011,696(2):572-580.
[4]GAMBAROTTA S,VAN BOLHUIS F,CHIANG M Y.Monomeric versus dimeric vanadium(iii)aryloxide formation.syntheses and crystal structures of[(O-2,6-ArMe2)2(μ-O-2,6-ArMe2)VIII]2·THF and[(O-2,6-ArMe2)3VIII(Py)2]2(O-2,6-ArMe2= 2,6-Dimethyphenoxide)[J].Inorganic Chemistry,1987,26(26):4301-4303.
[5]EDEMA J J H,MEETSMA A,GAMBAROTTA S,et al.Ligand and alkali metal cation control on the molecular complexity of anionic chromium(ii)aryloxides.preparation and crystal structure of dimeric(pho)10cr2li6(thf)6and(2,6-me2c6h3o)6cr2li2(thf)4and trimeric(guo)14cr3na9cl[guo =(ο-ch3o)c6h4o]with an encapsulated chloide ion[J].Inorganic Chemistry,1991,30(19):3639-3646.
[6]CAMPBELL C,BOTT S G,LARSEN R,et al.Preparation of titanium fluoroalkoxides by alcoholysis of titanium alkoxides[J].Inorganic Chemistry,1994,33(22):4950-4958.
[7]COFFINDAFFERTW,STEFFYBD,ROTHWELLIP,et al.Synthesis,structure,and bonding of mononuclear aryloxide derivatives of niobium in oxidation states+5, +3, +2,and+1[J].Journal of the American Chemical Society,1989,111(13):4742-4749.
[8]ARNEY D J,WEXLER P A,WIGLEY D E.Arene complexes of tantalum(iii)prepared by alkyne cyclization and alkoxide-exchange reactions.arene folding and π localization upon coordination[J].Organometallics,1990,9(4):1282-1289.
[9]GIESBRECHT G R,GORDON J C,BRADY J T,et al.Interactions of remote alkyl groups with lanthanide metal centers:synthesis,characterization and ligand redistribution reactions of heterobimetallic species containing trialkylaluminum fragments[J].European Journal of Inorganic Chemistry,2002,2002(3):723-731.
[10]ROTHWELL I P.Cyclometalation chemistry of aryl oxide ligation[J].Accounts of Chemical Research,1988,21(4):153-159.
[11]CHAMBERLAIN L,ROTHWELL I P,HUFFMAN J C.Intramolecular addition of an aliphatic ch bond to a tantalum-carbon double bond[J].Journal of the American Chemical Society,1982,104(25):7338-7340.[12]THORN M G,FANWICK P E,ROTHWELL I P.Reactivity of group 4 metal alkyl and metallacyclic compounds supported by aryloxide ligands toward organic isocyanides[J].Organometallics,1999,18(21):4442-4447.
[13]YU J S,FELTER L,POTYEN M C,et al.Intramolecular dehydrogenation of alkyl groups at niobium aryloxide centers:bonding and reactivity of the ensuing niobacyclopropane ring[J].Organometallics,1996,15(21):4443-4449.
[14]WOLCZANSKI P T.Structure and reactivity studies of transition metals ligated bytBuSi3X(X = O,NH,N,S,and CC)[J].Chemical Communications,2009(7):740-757.
[15]STANCIU C,OLMSTEAD M M,PHILLIPS A D,et al.Synthesis and characterization of the very bulky phenols Ar∗OH and Ar′OH(Ar∗ =C6H3-2,6-Trip2,Trip = C6H2-2,4,6-iPr3;Ar′= C6H3-2,6-Dipp2,Dipp= C6H3-2,6-iPr2)and Their Lithium and Sodium Derivatives(LiOAr′)2and(NaOAr∗)2[J].European Journal of Inorganic Chemistry,2003,2003(18):3495-3500.
[16]MALCOLMSON S J,MEEK S J,SATTELY E S,et al.Highly efficient molybdenum-based catalysts for enantioselective alkene metathesis[J].Nature,2008,456(7224):933-937.
[17]MEEK S J,O’BRIEN R V,LLAVERIA J,et al.Catalytic z-selective olefin cross-metathesis for natural product synthesis[J].Nature,2011,471(7339):461-466.
[18]HATANAKA T,MIYAKE R,ISHIDA Y,et al.Synthesis of two-coordinate iron aryloxides and their reactions with organic azide:intramolecular c-h bond amination[J].Journal of Organometallic Chemistry,2011,696(25):4046-4050.
[19]SEARLES K,TRAN B L,PINK M,et al.3d Early transition metal complexes supported by a new sterically demanding aryloxide ligand[J].Inorganic Chemistry,2013,52(19):11126-11135.
猜你喜欢
中学化学(2022年5期)2022-06-17
太原理工大学学报(2022年2期)2022-03-21
华南师范大学学报(自然科学版)(2021年4期)2021-08-30
化工环保(2021年3期)2021-06-17
无机化学学报(2020年7期)2020-07-20
理科考试研究·高中(2019年8期)2019-09-19
科技创新导报(2016年30期)2017-03-15
化学教学(2015年11期)2015-12-19
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
中国科技信息(2012年11期)2012-10-26
