
B、N共掺杂单层石墨烯电子结构和导电性能
2018-07-23 02:22任福成徐守冬刘世斌
太原理工大学学报 2018年4期
任福成,徐守冬,张 鼎,陈 良,刘世斌
(太原理工大学 洁净化工研究所,太原 030024)
近年来锂电池在生活中得到了很广泛的应用[1-4],但锂电池的能量密度尚不能达到汽油的能量密度(13 000 Wh/kg)[5]。为了使锂电池在未来能够应用在运输工具上,对锂电池的研究重点集中到发展新型锂电池体系,比如Li-O2[6-14]和Li-S[15-16]电池等。在这些新型电池中,Li-O2电池能量密度可达到11 682 Wh/kg,和化石燃料相当,因此具有很好的应用前景。Li-O2电池的高能量密度由电池负极金属锂和正极O2反应得到,O2可直接从空气中分离获取。然而Li-O2[17-18]电池的发展还处在初级阶段,推广应用受很多因素的限制,如电解液的稳定性不高、较高的反应过电势、低放电容量和循环性能差[19-21]等。提高电池的反应性能的常用方法之一就是采用电化学催化剂,如基碳类材料[22-23]、贵金属[24-25]、金属氧化物[26-27]和合金类催化剂[28]等;其中,基碳类催化剂因其大比表面积、高导电性和低成本等而受到广泛重视。在基碳类材料中,石墨烯因其质量密度轻(2 g/cm3)、比表面积大(2 630 m2/g)、化学稳定性高和金属性质的导电性而被广泛运用到Li-O2电池的正极材料中。KIM et al[29]将石墨烯作为负极材料,电池循环第1周和第10周的库伦效率分别为99%和87%.WU et al[30]合成的N掺杂石墨烯纳米片应用到Li-O2电池中,对ORR反应的催化性能与Pt/C催化剂相当。ZHAO et al[31]合成的3D多孔石墨烯气溶胶作为正极材料,在3.2 A·g-1下得到高比容量(5 978 mAh·g-1)和长循环寿命。
第一性原理研究方法在研究材料性能和反应机理方面有很大优势。JING et al[32]计算5种不同结构类型的N掺杂石墨烯,表明平面结构N掺杂石墨烯对ORR催化活性最好。REN et al[33]的研究表明,B掺杂石墨烯对锂电池中的析氧反应(OER)有促进作用,电池的充电速率明显提高。N掺杂石墨烯可作为ORR反应的一种高效催化剂,但是N掺杂比例和位置会不同程度地影响其带隙值。石墨氮掺杂的导电性随掺N量的增加而降低,而吡啶氮掺杂石墨烯(PNG)的导电性能随掺N量的增加先提高后降低[34]。LAREF et al[35]对不同结构类型的石墨烯分别进行B、N单元素掺杂,结果表明,B原子掺杂增强了石墨烯的导电性能,而N原子掺杂后其导电性能降低且随掺杂比例增加而减弱。ZHAO et al[36]的研究认为,B、N原子间位共掺杂活化了C原子的pz轨道电子,使石墨烯对ORR、OER反应有很好的催化作用;然而在Li原子存在的环境中,其催化效果并不明显。
电极材料的性能直接影响电池的应用。B、N单元素掺杂石墨烯作为具有催化性能的电极材料,研究者在其性能方面做了大量的研究,但是对掺杂后石墨烯的电荷分布、电子轨道杂化和轨道分布等研究很少。利用密度泛函理论研究B、N共掺杂石墨烯电子结构分布、导电性能以及共掺杂石墨烯材料在电池中的应用是很有必要的。本文对B、N原子以邻、间、对位共掺杂石墨烯的电子结构进行详细研究。B、N原子共同掺杂到一个碳环中,共掺杂模型和本征石墨烯是等电子体系,B、N共掺杂结构的电荷分布有明显的转移,共掺杂结构的能带和原子间的杂化类型较石墨烯发生了很大的变化,本文通过对能带、态密度和电子分布的分析来预测掺杂对材料性能影响。
1 计算方法和晶胞模型
1.1 计算方法
本次计算采用美国Accelerys公司开发的MS6.0(Materials Studio)软件中的CASTEP模块。几何优化和电子结构计算基于密度泛函理论(DFT)平面波赝势展开,采用广义梯度近似(GGA)修正的PBE泛函处理交换关联能[37-38],用超软赝势来描述价电子和离子实之间的相互作用。所有的计算都是在倒易空间中进行,几何优化过程中的算法都采用BFGS。本文计算平面波截断能(Energy cut-off)取为380 eV,第一布里渊的K-point设置为4×4×2,几何优化参数设置如下:迭代过程中的SCF收敛精度为2.0×10-5eV/atom,最大位移为2.0×10-4nm,原子间相互作用收敛标准设定为2.0×10-5eV/atom,内应力不超过0.1 GPa.为减小层与层之间的相互影响,真空度取2.5 nm.计算过程中的价电子组态分别为B:1s22s22p1,C:1s22s22p2,N:1s22s22p3.
1.2 晶胞模型
本文分别对晶胞模型为2×2×1石墨烯和B、N原子以邻、间、对位共掺杂石墨烯电子结构做详细研究,石墨烯结构如图1(a)所示。石墨烯结构中每个碳环由6个碳原子构成,每个碳原子被3个碳六环共用,所以石墨烯元胞有2个碳原子。图1(b)-图1(d)分别为B、N以邻、间、对位共掺杂石墨烯的结构示意图。B、N共掺杂石墨烯结构几何对称性降低,但3种共掺杂结构中B、C、N的原子比均为1∶6∶1(12.5%),B和N原子以相同比例掺杂石墨烯,保证掺杂结构和石墨烯是等电子体系,在等电子体系下研究B、N共掺杂对电子结构的影响,N掺杂量为7.54%和9%石墨烯对电池的循环和倍率性能有明显的改善[39-40]。3种结构的形成能分别为:邻位-2.31 eV,对位-1.20 eV,间位-1.14 eV。这一规律和JIANG et al[41]的结果一致,而且B、N邻位掺杂结构比石墨烯的二维结构还要稳定,所以这几种结构在反应过程中是稳定的[42-43]。
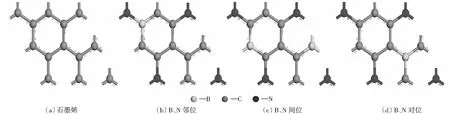
图1 共掺杂2×2×1石墨烯的结构模型图Fig.1 Schematic structure (top-view) of 2×2×1 graphene, B-NBG, S-NBG, P-BNG
2 结果与分析
2.1 能带结构
为讨论B和N以邻位(B-BNG)、间位(S-BNG)、对位(P-BNG)共掺杂对单层石墨烯的电子结构的影响,本次研究分别对单层石墨烯和共掺杂单层石墨烯的能带、态密度、电荷密度和电荷差分密度进行计算,能带结构如图2(a)-图2(d)所示。从石墨烯的能带结构分布(图2(a))看出,在费米能级附近价带最高点和导带最低点,在布里渊区H-K点之间相切可知,其能带分布具有金属性,有半金属性的导电性质[35]。如图 2(b)-图2(d)所示,B、N共掺杂对石墨烯的能带分布产生很大影响,价带和导带之间出现带隙,但都属于直接带隙。掺杂邻、间、对位的带隙值分别为2.406,1.296,2.572 eV;其中间位带隙最小,而且间位共掺杂结构的能带在布里渊区高对称点H-K(图2(c)),价带和导带同时出现最高点和最低点,与石墨烯相同。邻位和对位的带隙值接近,而且都在G点价带和导带发生分裂,如图2(b)和图2(d)所示,共掺杂形成的直接带隙半导体材料中间位共掺杂结构的带隙值最小,具有最好的导电性能。石墨N(GNG)掺杂比例为2.8%时,石墨烯能带就出现带隙,而且随着掺杂比例升高带隙增大,而且吡啶氮和吡咯氮对其带隙值得影响也不同[34]。

图2 共掺杂2×2×1石墨烯能带结构图Fig.2 Band structure of 2×2×1 graphene, B-NBG, S-NBG, P-BNG
共掺杂结构价带的能带不再简单的分布,能带增多而且能带之间的交叉更为复杂,表现为B、N共掺杂改变了石墨烯结构中原子间的杂化类型,掺杂后的结构中存在C—C,B—C,C—N和B—C—N等共同杂化。掺杂后导带的空轨道整体集中在2.5~7.5 eV,和石墨烯相比整体下移。B、N邻位、间位、对位共掺杂能带结构和石墨烯相比较的共同特点:价带能带数明显增多,而且更靠近费米能级,能带之间的交叉更为复杂,每条能带所占的能量区间减小,所有的能带结构较本征石墨烯变得平缓,电子轨道的扩展性减弱。LAREF et al[35]研究结果显示:B掺杂石墨烯形成P型石墨烯,价带能分布靠近费米能级,而N掺杂可得到N型石墨烯,导带能分布靠近费米能级,同样随着掺杂比例升高石墨烯能带结构中出现带隙。通过对共掺杂后特殊的能带结构分析可知,共掺杂结构在不同的环境下可作为很好的电子供体和受体。
2.2 态密度
图3(a)-图3(d)分别为石墨烯和B、N邻位、间位、对位共掺石墨烯态密度图。石墨烯态密度图(图3(a))费米能级两侧的峰,分别出现在-1.916,1.723 eV,主要来源于C原子2p轨道杂化的作用,两峰之间的能量差为3.639 eV.B、N原子共掺杂石墨烯的态密度发生明显变化,邻位共掺杂费米能级两侧的第一个峰分别出现在-0.579,2.287 eV;从图3(b)可以看出这两个峰属于B、C和N等3种原子的2p轨道共同杂化峰,能量差为2.866 eV.间位共掺杂费米能级两侧的第一个峰分别出现在-0.759,2.011 eV,杂化峰之间的能量差为2.760 eV,比邻位共掺杂石墨烯的能量差小0.106 eV;其中,费米能级左侧的第一个峰属于B、C原子2p轨道的杂化峰,费米能级右侧的第一个峰属于B、C、N三原子的共同杂化峰,而且B、C原子间的杂化作用更强。对位费米能级两侧的第一个峰分别出现在-0.208,2.472 eV,两个杂化峰之间的能量差为2.680 eV,比石墨烯的能量差小0.458 eV;其中,费米能级左侧的第一个峰属于B、C原子2p轨道的杂化峰,共掺杂结构在费米能级两侧态密度分布与石墨烯相比更靠近费米能级。N掺杂石墨烯其态密度在靠近费米能级的导带向低能级移动,B掺杂石墨烯则相反价带向高能级移动。计算结果表明,B和N共掺杂结构的价电子轨道和空轨道同时靠近费米能级,这一结果很好地结合了B和N单元素掺杂的特点[32-35]。对比图3(a)-图3(d)中费米能级两侧第一个峰,间位掺杂态密度的峰值和峰宽最大,表明共掺杂中间位可提供更多发生跃迁的电子,同时导带底可为外电子提供更多空轨道。共掺杂结构中C—N原子杂化轨道分布在低能量区域,B—C原子杂化轨道分布更靠近费米能级,在-20 eV附近均存在一个C—N原子2s轨道的杂化峰。通过对能带结构和态密度的分析可知,B、N共掺杂中间位共掺杂结构的带隙最小,导电性能最优,化学活性也最强。
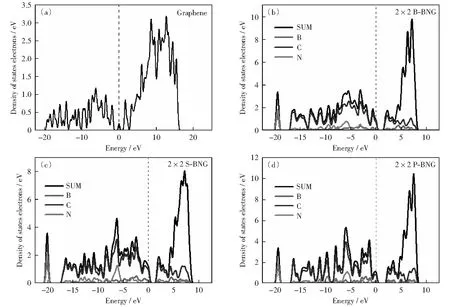
图3 石墨烯,B、N邻位,B、N间位,B、N对位共掺杂2×2×1石墨烯态密度图Fig.3 Density of states of 2×2×1 graphene, B-NBG, S-NBG, P-BNG
2.3 间位共掺杂结构原子电荷布局分析
石墨烯的二维结构中所有C原子之间为sp2杂化,在C原子的2s和2p轨道分别分布1.05e和2.95e电子,pz轨道的电子形成π电子对。由于B、C、N等3种原子之间电负性的差异:B(2.04e),C(2.55e),N(3.04e),B和N原子间位取代两个C原子后体系中N原子显负电性,B原子显正电性,C原子电荷分布平衡被打破,电荷分布发生偏移。从原子电荷布局分析(表1)可知,与B、N原子都成键的C2原子,从B原子得到的电子数比C2贡献给N原子电子多0.1e;体系电荷重新分布后,B原子失去0.55e电子,比邻位掺杂失去电子数少0.16e;2s和2p轨道上分别分布0.54e和1.91e电子,2p轨道上的电子在与C2,C4,C5的成键中起主要作用。与B原子成键的C5原子表现出最大的负电性,其2s和2p轨道分别得到0.09e和0.16e电子,N原子2s和2p轨道上分别分布1.41e和3.80e电子,得到0.21e电子。与N原子成键的C6原子,表现出最大的正电性,其2s轨道失去0.01e电子,2p轨道失去0.12e电子,二维结构中与B、N原子距离越大,C原子的电荷转移数越小。通过间位共掺杂各原子2s和2p轨道电荷转移情况的分析可知,与结构中B原子对C原子电荷转移的影响相比,N原子的影响更为明显;各原子的电荷转移主要发生在原子的2p轨道,其原因是2p为B,C,N原子最外层电子轨道,B,N原子取代石墨烯结构中的C原子最先影响分布在原子最外层轨道上的电子。对比B,N邻、间、对位共掺杂结构中C原子电荷转移量可知,间位掺杂结构中C原子的电荷转移量均小于邻和对位掺杂,其中B原子的电荷转移量由小到大为:间位(0.55e),对位(0.61e),邻位(0.71e);N原子的电荷转移量由小到大为:间位(-0.21e),对位(-0.25e),邻位(-0.43e).
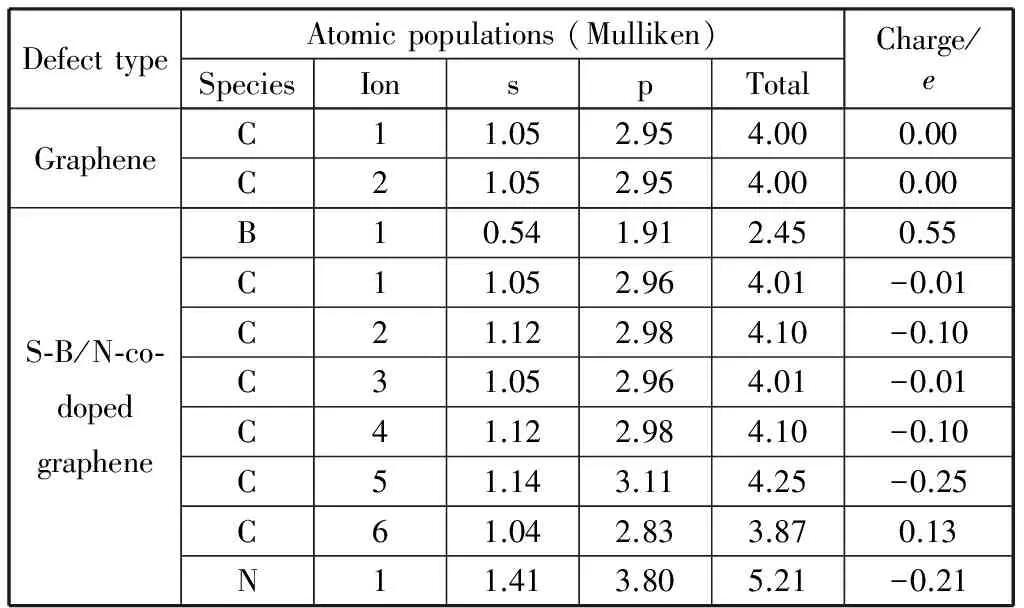
表1 石墨烯和B、N间位共掺杂2×2×1石墨烯原子电荷布局分析Table 1 Atom Mulliken-poppulation analysis of 2×2×1 Graphene and S-BNG
2.4 间位共掺杂结构电荷密度和电荷差分密度

图4 石墨烯和B、N间位共掺杂石墨烯电荷密度图Fig.4 Electron density contour of graphene and S-NBG
图5为B、N间位共掺杂石墨烯的电荷差分密度图,图中蓝色区域和红色区域分别表示电荷密度减小(Δρ<0)和增大(Δρ>0).电荷差分密度Δρ计算公式:
Δρ=ρTotal-ρGraphene-∑nρi.
式中:ρTotal是掺杂体系优化后的总电荷密度分布函数;ρGraphene为石墨烯体系的电荷密度分布函数;ρi为B、N原子的电荷密度分布函数;n为B、N原子的掺杂数。在图中的电荷密度值为0.017 4~0.716 8的区间选取了两个电荷密度相等的面,其密度数值分别为0.273,0.443 .从等电荷密度面的分布可以看出,掺杂后电子云发生偏移,π电子对被破坏,等密度面不再沿着六边形均匀分布,发生很大偏移。电荷密度为0.273的等密度面在B和C原子之间的偏移量很小,但是在C和N原子之间发生了明显的偏移,更加靠近N原子;0.443的等密度面在B和C原子之间发生了偏移,偏离B原子靠近C原子,同样,在C和N原子之间的分布也更靠近于N原子。在没有和B、N原子成键的C原子之间,两个等势面均匀地分布在两个C原子之间[45],掺杂之后材料的极性由非极性向极性转变。
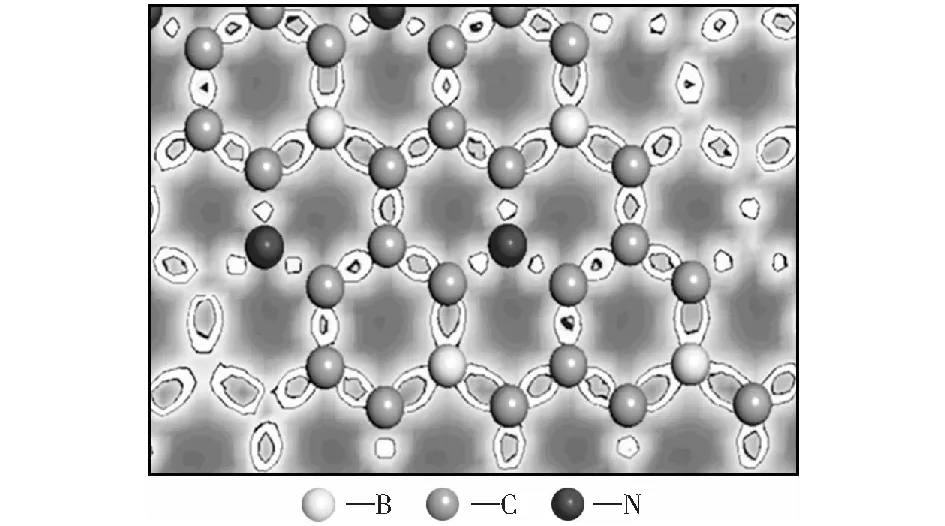
图5 B、N间位掺杂石墨烯电荷差分密度图Fig.5 Electron density difference of S-BNG
2.5 HOMO轨道
图6为B、N原子间位共掺杂石墨烯的HOMO轨道(最高分子占据轨道)分布图,能量范围为-1.541 4~0.000 0 eV.理论上,HOMO电子的轨道和LUMO轨道(最低未占分子轨道)的能量差最小,即电子吸收能量跃迁到导带空轨道的能量差最小。从图6可以看出,最高占据态轨道主要分布在B、C原子周围,N原子周围只有很小一部分。B、N共掺杂2×2×1石墨烯结构的表面被活化,B和C原子的价电子更容易发生跃迁。
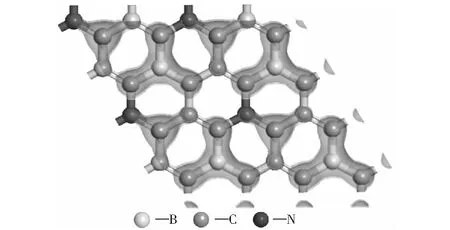
图6 B、N间位共掺杂石墨烯HOMO轨道分布图Fig.6 HOMO distribution of S-BNG
3 结论
本文利用密度泛函理论计算研究了B、N原子以邻、间、对等3种位置共掺杂石墨烯结构的电子结构,详细分析了B、N原子掺杂对石墨烯电子分布和导电性能的影响,以及B、C和N等3种原子之间的互相杂化关系,得出以下的结论。
1) 在B、N掺杂比例同为12.5%的条件下,共掺杂结构的能带出现带隙,都属于直接带隙;其中,间位的带隙值最小(1.296 eV),而且间位掺杂结构和石墨烯的能带结构中价带最高点和导带最低点相同,间位导电性能最好。
2) B、N共掺杂石墨烯的价电子轨道和空轨道分布更靠近费米能级,且B,C,N等3原子之间互相发生杂化和共同杂化,其杂化主要是各原子的2p轨道之间的杂化作用。其中,B、C原子价电子杂化轨道所占据的能量较高,靠近费米能级;B、C和N原子的价电子杂化轨道分布在低能量状态;而C和N原子的空轨道分布更靠近费米能级。
3) B、N间位共掺杂结构中各原子的电荷转移量最小,对石墨烯电子结构的影响最小,B原子失去0.55e电子,N原子得到0.21e电子。共掺杂结构表面被活化,分布在B和C原子周围的价电子更容易发生跃迁,而N原子所在区域更容易接收外来电子。
猜你喜欢
无机化学学报(2022年9期)2022-09-16
井冈山大学学报(自然科学版)(2022年2期)2022-03-31
小天使·聪聪画刊(2021年2期)2021-09-10
中外文摘(2021年7期)2021-04-23
汽车零部件(2020年10期)2020-11-09
发明与创新·小学生(2020年10期)2020-10-19
发明与创新·小学生(2019年12期)2019-12-05
汉语世界(The World of Chinese)(2019年6期)2019-09-10
汽车生活(2018年5期)2018-06-21
读写算·教研版(2016年8期)2016-05-07
