
锡烯与一些亲核试剂加成反应的理论研究
2018-08-10 09:32曾小兰
信阳师范学院学报(自然科学版) 2018年3期
曾小兰,王 岩
(信阳师范学院 化学化工学院,河南 信阳 464000)
0 引言
包含C=C双键的烯烃是一类重要的有机化合物.包含M=C双键(M = Si, Ge, Sn, Pb)的化合物称为金属烯.BROOK等[1]在1981年首次制备得到包含Si=C双键的硅烯.自那以后,有关各种金属烯的结构、性质和反应性的实验研究层出不穷.BERNDT等[2]在1987年合成了第一个包含Sn=C双键的锡烯化合物.ANSELME等[3,4]在随后的研究中发现,包含Sn=C双键的锡烯室温下很容易与各种亲核试剂如H2O、MeOH、PhNH2及HCl发生加成反应且亲核试剂中带负电荷的O、N、Cl原子无一例外地加成到Sn原子上.
与此同时,理论化学家也对金属烯化合物的结构、性质及反应性倾注了极大的兴趣,但有关的理论计算研究主要集中在硅烯与两种亲核试剂H2O及MeOH之间的加成反应[5,6].计算结果显示,硅烯的动力学稳定性与Si=C双键中Si原子和C原子上的取代基的性质密切相关.直到近期,LI等[7]采用密度泛函理论(DFT)方法研究了包含锡烯在内的金刚烷取代的金属烯化合物与MeOH的单聚体及二聚体之间的加成反应的微观机理和势能剖面.但他们只考虑了一种亲核试剂,也没有探讨M=C双键(M = Si, Ge, Sn, Pb)中M原子和C原子上的取代基的影响.
本文采用DFT方法研究了锡烯与三种不同的亲核试剂(H2O、MeOH及PhOH)间形成Sn—O和C—H键的加成反应的微观机理和势能剖面,比较了几种亲核试剂反应活性的高低,分析了Sn=C双键中Sn原子和C原子上的取代基对反应势能剖面的影响.本研究工作将有助于深入了解相关金属烯化合物的结构、性质和反应性.本文所研究的相关反应见图1.

图1 锡烯与亲核试剂的加成反应Fig. 1 The addition reactions betweenstannenes and nucleophilic reagents
1 计算方法
作为最广泛使用的DFT方法之一,B3LYP方法[8,9]的可靠性在先前的计算化学研究[7,10,11]中被证实.本文采用B3LYP方法,对C、H、O及Si原子使用6-311++G(d,p)基组,对Sn原子使用Lanl2dz基组,全优化计算所研究反应的反应物、分子复合物、过渡态及最终产物的分子几何构型.通过振动频率计算,以证实所优化得到的构型为势能剖面上的能量极小点(反应物、分子复合物及产物)或鞍点(过渡态).根据过渡态的虚振动模式判断它们与相邻的两个极小点的正确连接.各分子中的原子电荷使用自然键轨道(NBO)方法计算得到.本文的全部计算均使用Gaussian 03程序.
2 结果与讨论
本文所涉及的锡烯与三种亲核试剂(H2O、MeOH及PhOH)的加成反应共有11个(如图1所示).为便于讨论,这些反应用符号N(N = 1~11)表示,其中的分子复合物(C)、过渡态(T)和最终产物(P)则分别用CN、TN及PN表示.
2.1 反应机理
研究结果显示,H2O及MeOH与锡烯的母体化合物H2Sn=CH2之间的加成反应都可能以三种方式进行.具体而言,H2O及MeOH的单聚体、二聚体及三聚体可分别与H2Sn=CH2发生加成反应(图1中的反应1~3或4~6).而且还发现,反应1、2及3的机理与反应4、5及6基本相同.因此,本文主要考虑反应1~3的机理.
在H2O的单聚体与H2Sn=CH2之间的加成反应1中,两个反应物分子首先形成一分子复合物,然后经过协同的四元环过渡态变成最终产物.该反应的机理与以前报道的H2O单聚体与硅苯-Cr(CO)3配合物的1,2-加成机理[10]很相似.图2呈现了反应1及反应2所涉及的几个分子复合物、过渡态及最终产物的分子结构示意图及与反应有关的关键原子的编号,各相关键的键长(nm)也一并列出.
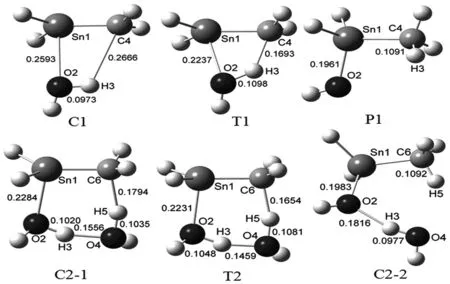
图2 与反应1及反应2有关的分子间复合物、过渡态及产物的分子结构示意图、原子编号及相关键的键长(nm)Fig. 2 Molecular structures of complexes (C), transition states (T), and products (P) for reactions 1 and 2 along with the numberingsystem for some key atoms, indicating relevant bond lengths (nm)
从图2可以发现,在反应1的初始分子复合物C1中,Sn1—O2间距离(0.2593 nm)比最终产物(P1)中的Sn—O单键的键长长大约0.063 nm,说明在C1中只存在弱的Sn1—O2相互作用.而C1中C4—H3间距离(0.2666 nm)远大于P1中的C—H单键的键长,说明在C1中基本不存在C4—H3相互作用.另外,C1中O2—H3间距离(0.0973 nm)只比孤立水分子中的O—H键键长略微长一些.由C1中各原子的NBO电荷可发现,C1形成时有大约0.085 e电荷从水分子转移到H2Sn=CH2分子,说明C1是一个典型的亲核复合物.
在反应1的过渡态T1中,二面角C4H3O2Sn1(5.03)非常接近于0,说明C4、H3、O2及Sn1四个原子基本在同一个平面上,或者说T1为一平面四元环过渡态.T1中Sn1—O2间距离只比P1中长0.0276 nm,而C4—H3间距离比P1中长0.0602 nm,说明在反应1中Sn—O键比C—H键更先形成.
反应2及反应3的机理与反应1大致相同,H2O的二聚体或三聚体与H2Sn=CH2也首先形成一初始复合物(C2-1或C3-1),然后经过六元环或八元环过渡态(TS)形成第二个复合物(C2-2或C3-2).H2O的单聚体或二聚体从C2-2或C3-2中脱去便得到P1.
从图2可以发现,C2-1中的Sn1—O2键长比C1中的短大约0.031 nm,说明前者中的Sn1—O2相互作用强于后者;同时,C2-1中的C6—H5键长(0.1794 nm)明显短于C1中的C4—H3距离,说明在C2-1中已有较强的C6—H5相互作用.对于反应2,从C2-1经过T2到C2-2,Sn1—O2键和C6—H5键逐渐形成,O2—H3键和O4—H5键逐渐断开,同时形成一个新的O—H键,即O4—H3键.需要注意的是,在C2-2中与P1通过O—H…O氢键相结合的H2O单聚体并不是作为反应物的H2O二聚体中的某一个H2O分子,而是两个H2O分子分别提供OH基团和H原子形成的.这说明在反应2中,H2O二聚体中的一个H2O分子实际上起到传递质子的桥梁作用.反应3的情况非常相似,H2O三聚体中的两个H2O分子也起到传递质子的作用.
对于PhOH与H2Sn=CH2之间的加成反应,计算发现,只能优化得到PhOH单聚体作为亲核试剂的反应的过渡态,而未能优化出PhOH二聚体及三聚体作为亲核试剂的反应的过渡态.这可能是因为,Ph基团有较大的体积,导致在PhOH二聚体及三聚体作为亲核试剂的反应中有很大的空间位阻效应.
2.2 反应的势能剖面
以两反应物在298.15 K时的Gibbs自由能之和作为能量零点计算得到了各反应中分子复合物、过渡态及产物的相对Gibbs自由能Gr.对于最终产物,其Gr即为相应反应的反应Gibbs自由能(rG);而过渡态的Gr即为对应反应的活化Gibbs自由能(rG).考虑到文章的篇幅.图3只列出了反应1、反应2及反应3的自由能势能面图.
根据图1,反应1、反应2及反应3在热力学上是等价的,因此这三个反应的rG完全相同.同理,反应4、反应5及反应6的rG也完全相同.从图3可以看出,反应1、反应2及反应3的rG为196.4 kJ/mol,说明室温下这些反应为热力学自发过程,或者说他们在热力学上很容易进行.但这些反应的动力学行为并不相同.由图3可以发现,反应1、反应2及反应3的rG分别为35.2、7.0及5.2 kJ/mol,说明反应3即H2O三聚体与母体锡烯H2Sn=CH2的加成反应在动力学上最容易进行.仔细分析表明,反应2的rG远低于反应1主要起源于能量因素,因为六元环过渡态的张力小于四元环过渡态且前者中存在O—H…O氢键相互作用.而反应2和反应3的rG比较接近是能量因素和熵因素共同作用的结果.需要说明的是,尽管T1的Gr高于C1,但T2或T3的Gr反而分别低于C2-1或C3-1(见图3),这是因为T2或T3的零点振动能分别低于C2-1或C3-1.

图3 反应1、反应2及反应3的Gibbs自由能势能剖面图Fig. 3 Gibbs free energy profiles of the reactions 1, 2 and 3
为了探讨亲核试剂的种类对加成反应的影响,本文还考虑了PhOH作为亲核试剂的可能加成反应.如前面所讨论的,我们只能优化得到PhOH单聚体作为亲核试剂的反应(图1中的反应7)的过渡态,计算得到的该反应的rG为39.3 kJ/mol,说明PhOH作为亲核试剂的反应活性低于H2O和MeOH.我们认为,这很可能是由于Ph基团有较大的空间位阻效应造成的.
2.3 反应的取代基效应
为了研究Sn=C双键中Sn原子和C原子上的取代基对反应势能剖面的影响,本文还考虑了图1中的反应8~11.这四个反应的机理与反应1非常相似,计算得到的rG分别为29.3、59.2、46.7及34.1 kJ/mol.与反应1的rG(35.2 kJ/mol)比较可以看出,Sn=C双键中的Sn原子上的吸电子基团Ph或者C原子上的给电子基团SiH3能降低反应的活化Gibbs自由能,或者说Sn原子上的吸电子基团Ph或者C原子上的给电子基团SiH3使反应在动力学上变得更有利些.另一方面,Sn=C双键中的Sn原子上的给电子基团SiH3或者C原子上的吸电子基团Ph能使反应的活化Gibbs自由能升高,或者说使反应在动力学上变得不利.上述结果可能与Sn=C双键极性的变化有关.在本文所涉及的五种锡烯(H2Sn=CH2、Ph2Sn=CH2、H2Sn=CPh2、(SiH3)2Sn=CH2及H2Sn=C(SiH3)2)中,Sn=C双键中的Sn原子都携带正电荷,而其中的C原子都携带负电荷.可以用Sn原子与C原子所携带的电荷的差值(t)来表征Sn=C双键的极性,t越大,Sn=C双键的极性越强.NBO分析表明,上述五种锡烯的t分别为2.363、2.999、1.706、1.995及3.203 e.可以看出,Sn原子上的吸电子基团Ph或者C原子上的给电子基团SiH3使Sn=C双键的极性增大,因此有利于亲核加成反应的进行;而Sn原子上的给电子基团SiH3或者C原子上的吸电子基团Ph使Sn=C双键的极性减小,不利于亲核加成反应的进行.
3 结论
本文采用DFT中的B3LYP方法,结合6-311++G(d,p)基组(对Sn原子使用Lanl2dz基组),研究了锡烯与一些亲核试剂(H2O、MeOH及PhOH)间的加成反应的微观机理、势能剖面及取代基效应,得到以下结论.锡烯与H2O或MeOH的单聚体、二聚体及三聚体均可发生反应,但其只能与PhOH的单聚体发生反应.锡烯与H2O或MeOH的三聚体间的反应在动力学上比其与相应的单聚体和二聚体间的反应有利.在本文所涉及的三种亲核试剂中,MeOH的反应活性最强,PhOH的反应活性最弱.Sn=C双键中Sn原子上的吸电子基团以及C原子上的给电子基团使反应在动力学上更有利,而Sn原子上的给电子基团以及C原子上的吸电子基团则使反应在动力学上变得不利.锡烯参与亲核加成反应的活性与Sn=C双键的极性密切相关.本研究结果对深入了解相关金属烯化合物的结构、性质和反应性有重要启示.
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06
油气·石油与天然气科学(2021年12期)2021-12-11
合成树脂及塑料(2020年4期)2020-09-20
新课程·下旬(2019年7期)2019-09-17
铜仁学院学报(2018年6期)2018-07-05
电脑知识与技术(2018年3期)2018-03-21
分析化学(2017年12期)2017-12-25
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
