
以N^N为辅助配体的铱配合物的电子结构和光谱性质的理论研究
2018-12-27 06:00曲晓春
世界有色金属 2018年20期
曲晓春
(延边大学理学院,吉林 延吉 133000)
作为新一代平面显示器的有机电致发光器件(OLED)具有视角广、画质均匀、图像稳定、反应速度快、较易彩色化、分辨率高、用简单驱动电路即可达到发光、制作过程简单,可实现柔性显示等优点。目前可作为有机电致发光器件的材料很多,其中铱配合物是目前研究的最多、也是最有应用前景的一类磷光电致发光材料。
首先,它具有强烈的自旋-轨道耦合,理论上电致发光器件内部量子产率可达到100%,其次,它的发射波长和效率具有可调性,室温下可实现磷光发射,因此铱配合物能广泛应用于有机电致发光领域。然而OLED还存在材料、基板技术、彩色化技术等方面问题,因此铱配合物在配体的选择上还需不断改进和创新,才能使OLED的不足得到改善。本研究以2-苯基吡啶(ppy)为主配体,分别以2-苯基-1,10-邻菲罗啉(N^N1)和8-苯基-3H-吡唑并[4,3-h]喹啉(N^N2)为辅助配体的两个金属铱配合物,通过改变辅助配体来研究比较这两个铱配合物的分子结构、电子结构和光谱性质,以阐明不同N^N辅助配体对配合物的影响。
1 计算方法
本文的计算均采用密度泛函理论。C、H、N原子的计算采用6-31G*基组[1];Ir原子则采用“double-ζ”的LANL2DZ赝势基组[2],其内层电子用有效核势进行了有效代替,外层电子5s25p65d6作为价电子。有效核势是一种对内层电子进行有效代替的近似方法。文中分子的最低单重态(S0)和三重态(T1)构型分别采用了DFT/B3LYP方法和非限制性的DFT/B3LYP(UB3LYP)方法进行了优化,并用同样的方法基组进行了频率计算。频率计算表明,所有优化得到的几何构型均没有虚频。在基态和激发态优化的基础上,用TDDFT方法和极化连续介质模型(PCM)和CH3CN溶剂分别计算了吸收和发射光谱。
2 结果与讨论
2.1 基态和激发态的分子结构
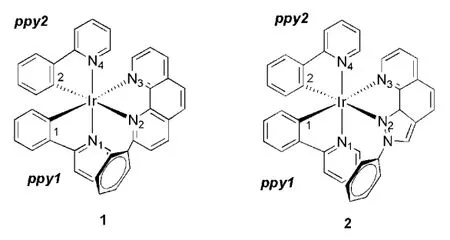
图1 分子平面图及标号
为得到性能优良的红光材料,根据已报道的绿光配合物1,改变辅助配体设计了配合物2,分子结构如图1所示,所需的各原子标号也标在图1中。配合物1与2的基态和激发态键长键角及差值数据及配合物1的实验数据列于表1。由表1中数据可知,除了Ir-N2键外,1中与铱相邻各原子的键长差值均在0.012 Å~0.066 Å,计算精确度在可以接受范围,而Ir-N2键计算值比实验值多0.153Å,这可能因为Ir-N2键受辅助配体上的苯环影响,在晶体结构中配合物的辅助配体上的苯环存在着堆积作用。配合物1的优化键长结果与实验值相对吻合。在键角比较中发现,1的计算值与实验值吻合较好,差值在0.08°~1.24°。键角N3-Ir-C1和N1-Ir-N4相对更接近180°,键角N2-Ir-C2是169.7°,弯曲程度最大,所以说N^N配体对含C2的ppy2配体的引力较大。

表1 配合物1与2的基态和激发态键长键角及差值数据及配合物1的实验数据

表2 配合物1和2在基态的分子轨道组成(%)和能级(eV)
通过1的基态和激发态分子的键长差值可以看出,其差值范围很小都低于0.05Å,而最大的差值来自于Ir-N2键长,激发态键长比基态短0.156Å。同理2的基态和激发态分子键长在数值上偏差也较小,最大的差值也是Ir-N2键长为0.087Å。键角方面,1的基态基本上比激发态对应的键角小1°左右,而基态的键角N2-Ir-C2反而比激发态大5.82°,差别最为明显。基态和激发态分子虽分子结构相似,但微观上仍具有较多差异。对于晶体和基态的配合物1和2,它们的Ir-C1均比Ir-C2长的原因可能是:在配合物中,N^N辅助配体中悬挂的苯环和ppy1配体中的苯环形成了分子内面面间的π-π堆积作用。
2.2 电子结构
材料的光学性质与其前线分子轨道相关,其中分子最低空轨道(LUMO)和最高占据轨道(HOMO)尤为主要。表2列出了1和2的能级及轨道分布。从表2可以看出,1的HOMO轨道主要分布在ppy1配体的苯环上及Ir中心上,ppy2上只有很少比例。2的HOMO轨道分布多数也在ppy1和Ir上,但是其ppy2上分布的比例大于配合物1中ppy2的比例。1和2的区别在于1中N^N配体上的一个吡啶环变为吡唑环,由此推断,该含吡唑环的N^N配体对HOMO轨道在ppy配体的分布具有一定的均衡作用。1的LUMO轨道主要分布在辅助配体N^N1上,且比例相对平均,Ir上几乎没有。2中也是主要分布在N^N2上,1和2的LUMO轨道成分是极其相似的。
1和2的N^N辅助配体不同,其HOMO,LUMO的能级水平不同,其HOMO-LUMO的能隙也相差较大,分别为2.92eV和3.33eV,但是HOMO水平相似,分别为-7.70eV和-7.75eV,2的LUMO轨道更高些,这意味着由1到2,辅助配体引入的原子不同,改变的主要是分子的LUMO能级水平,继而改变了能隙的较大差异。
2.3 激发态能量和吸收光谱
配合物1和2的吸收光谱如图2所示。从吸收带的位置可以看出,1和2的峰值位置相差不多,2的峰值在279nm处,相对于1的262nm发生了红移。这表明吸收电子基团-CN 的引入,对此类材料的光物理性质有着较大的影响。2的最高吸收峰要高于1的,这就可能会增加单重激发态到三重激发态的系间窜越的可能性,从而增加量子效率。另外,1的第二吸收峰位于307nm,2的第二吸收峰位于326nm,且1的第二吸收峰高于2的。1的主要S0→S1跃迁是483nm,要大于2的424nm的波长。

图2 配合物1和2的吸收光谱
2.4 磷光发射光谱
为了在TDDFT泛函中选择合适的计算方法,本文分别用B3LYP、M062X及cam-B3LYP方法计算配合物1和2的磷光发射波长。
计算结果表明,使用cam-B3LYP方法计算得到配合物1的磷光发射波长为511.7nm,与Rubén D. Costa[3]等人报道的530nm相差最小,只有18.3nm。而采用B3LYP、M062X方法计算得到的分别为642.3nm和426.5nm,与文献值相差100nm左右,偏差较大。
所以对此体系采用cam-B3LYP方法计算比较准确,下面就是对用该方法计算的发射光谱性质进行讨论。
1的发射光谱的跃迁主要是由H-8→L+1和H-3→L+1跃迁组成的,各轨道跃迁成分较小,相应的电子跃迁特征为[d(Ir)+ π(ppy) →π*(N^N)],属于金属到配体的电荷转移和配体到配体的电荷转移(3MLCT/3LLCT)。
而2的发射光谱的跃迁来自HOMO-1→LUMO和HOMO→LUMO跃迁,跃迁性质为金属到配体的电荷转移和配体到配体的电荷转移以及配体内的电荷转移(3MLCT/3LLCT/3ILCT)。
计算得到2的磷光发射波长为589.7nm,与1的511.7nm相比,2发生了显著的红移,红移了78nm。
3 结论
本文运用密度泛函理论对两个铱配合物的基态和激发态分子结构、分子轨道、吸收和发射光谱进行了计算。计算结果表明,在配合物的键长和键角比较方面,与Ir-N2相关的键长和键角均出现较为明显的差异性;1和2的HOMO能量水平相似,而2的LUMO水平比1的高;2的磷光发射波长为589.7nm,与1相比,2红移了78nm,紅移显著。
根据以上计算结果可知,不同的辅助配体可以对金属铱配合物的光物理性质产生重要影响,我们推断配合物2有可能成为更好的红光磷光发光材料。
猜你喜欢
高中数理化(2022年16期)2022-09-14
内燃机工程(2021年1期)2021-02-05
汕头大学学报(自然科学版)(2020年4期)2020-12-14
青岛大学学报(工程技术版)(2019年2期)2019-09-10
中学生数理化(高中版.高考理化)(2019年6期)2019-06-22
分析化学(2017年12期)2017-12-25
分析化学(2017年12期)2017-12-25
东西南北(2016年14期)2016-08-16
原子与分子物理学报(2015年3期)2015-11-24
读写算·教研版(2014年12期)2014-09-01
