
3-氯-5-(三氟甲基)-2-乙胺基吡啶的合成
2019-04-01 09:49孙天孜杜友兴
有机氟工业 2019年1期
孙天孜 何 立 杜友兴
(1.上海威耳化工科技有限公司,上海 200331; 2.上海康鹏科技有限公司,上海 200331)
0 前言
随着现代农业的发展,杀菌剂特别是含氟酰胺类杀菌剂[1-2]得到了更加广泛的应用。3-氯-5-(三氟甲基)-2-乙胺基吡啶是制备酰胺类杀菌剂氟吡菌酰胺的重要中间体,具有杀菌谱广、高效低毒等特点,研究其合成具有重要的现实意义。其合成工艺主要有以下几种:1)以3-氯-5-(三氟甲基)-2-甲醛基吡啶作为起始原料,经肟化、脱肟和氢化还原3步反应制备,该工艺起始原料难以得到,不适宜工业化生产[3-7]; 2)以二苯甲酮作为起始原料,经取代反应和脱羧反应制备[8-9],该工艺需用到价格较贵的溶剂,且收率较低,操作复杂,生产成本较高,也不适宜工业化生产;3)以2,3-二氯-5-三氟甲基吡啶为起始原料,经亲核取代、脱羧、加氢还原和酰胺水解等4步反应制备[10-11],但在该工艺中亲核取代和脱羧反应是分别进行的,并且亲核取代反应结束后要加入盐酸和水以淬灭反应,收率较低,产生三废较多,且反应溶剂N,N-二甲基甲酰胺(DMF)不能直接用于下一批次的反应。脱羧这一步反应要加入DMF和盐酸,也会产生大量的“三废”。
为了解决上述工艺操作复杂、“三废”较多的问题,开发了亲核取代反应和脱羧反应“一锅法”进行的工艺,采用该工艺,反应溶剂可直接回收和重复利用,无废水产生,且两步反应收率比文献[10]报道的收率高7%。根据还原和水解两步反应的杂质定性结果,对还原和水解两步反应进行了优化,整个工艺合成步骤如图1所示。
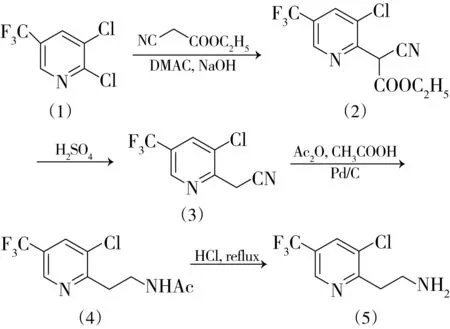
图1 3-氯-5-(三氟甲基)-2-乙胺基吡啶的合成工艺
1 试验部分
1.1 试验原料与仪器
2,3-二氯-5-三氟甲基吡啶,江苏威耳化工有限公司,质量分数为98.2%;氰乙酸乙酯,上海化学试剂公司。其他原料均为商业可得的国产工业级产品,未经处理直接使用。
HP6890/5973MSD型气相-质谱联用仪,美国HP公司,EI离子源;Agilent 1200/6220液相色谱-质谱联用仪,美国安捷伦公司;安捷伦1200系列液相色谱仪,美国安捷伦公司;岛津GC-2014C气相色谱仪,日本岛津公司,毛细管柱;Advance DMX400型核磁共振仪,四甲基硅烷(TMS)为内标,德国Bruker公司。
1.2 3-氯-5-(三氟甲基)-2-乙腈基吡啶(3)的合成
在2 L四口圆底烧瓶中,依次加入N,N-二甲基乙酰胺(DMAC)864.0 g、氢氧化钠52.0 g(1.3 mol)和2,3-二氯-5-三氟甲基吡啶 216.0 g(1.0 mol),60~70 ℃滴加氰乙酸乙酯147.1 g(1.3 mol)。滴加完毕后,60~70 ℃反应3 h。然后控制内温为110~120 ℃滴加浓硫酸,滴加过程中注意控制体系pH=1~2。滴加完毕,110~120 ℃保温反应3 h。降温至室温,过滤,用少量DMAC漂洗滤饼,滤液先用水泵减压回收DMAC,回收的DMAC可直接用作下一批次反应的溶剂,再用油泵减压收集196.2 g浅黄色固体化合物3,气相色谱测得其质量分数为98.6%,收率88.0%,沸点98.0~100.0 ℃(500 Pa)。核磁共振氢谱(1H NMR)(CDCl3,300 MHz)δ:8.95~8.65 (m,1 H),8.15~7.92(m,1 H),4.31~3.97(m,2 H);核磁共振碳谱(13C NMR)(CDCl3,300 MHz):17.8,117.8,122.4,123.6,124.1,130.5,133.8,134.4,136.4;气相色谱-质谱(GC-MS)m/z(%):219.01(100),221.00(32.0),220.01(9.8),222.01(3.1)。
1.3 3-氯-5-(三氟甲基)-2-吡啶基乙基乙酰胺(4)的合成
向哈氏合金高压釜中依次加入60.0 g(0.273 mol)化合物3、乙酸360 g、乙酸酐110.2 g(1.092 mol)及质量分数为5%的Pd/C 9.0 g,用N2置换3次,然后于3.0 MPa、20 ℃条件下反应12 h。过滤,将滤液减压旋蒸脱除溶剂得到98.0 g淡黄色固体化合物4,经高效液相色谱(HPLC)测得其纯度为87.5%,可直接用于下步反应。
1.4 3-氯-5-(三氟甲基)-2-乙胺基吡啶 (5)的合成
在1 L四口瓶中加入水300.0 g、质量分数为30%的浓盐酸166.1 g(1.365 mol),然后加入98.0 g(0.273 mol)化合物4,在搅拌下回流反应8 h。冷却至室温,反应液用二氯甲烷(3×50 g )萃取,得到的水相先用质量分数30%的氢氧化钠溶液调节pH=8~9,再用二氯甲烷(3×60 g )萃取。合并二氯甲烷相,短蒸后得到53.9 g浅黄色油状液体化合物5,外标含量98.2%,收率79.9%。4步反应总收率为68.8%。1H NMR(CDCl3,400 MHz)δ:8.71(s,1H),7.98~7.84(m,1H),3.23~3.17(m,2H),3.15(dd,J=9.6 Hz,3.8 Hz,2H),1.52(s,2H);13C NMR(CDCl3,400 MHz):31.9,39.2,123.6,127.1,130.8,136.1,147.7,160.7;GC-MS m/z(%):224.03(100.0),226.03(32.0),225.04(8.7)。
2 结果与讨论
2.1 化合物3的合成工艺条件优化
化合物3的“一锅法”合成涉及亲核取代和脱羧两步反应。亲核取代反应需要在碱性条件和一定温度下的极性溶剂中进行,主要考察了反应溶剂、原料配比、碱的种类和数量对亲核取代反应收率的影响,而脱羧反应需要在酸性条件、较高温度下进行,主要考察了反应体系的pH和反应温度对水解脱羧反应的影响。在研究过程中通过气相色谱(GC)确定反应进程和产品含量,通过高压液相色谱(HPLC) 测定反应液的外标含量进而计算反应收率。
2.1.1碱的种类和用量对化合物2收率的影响
以N,N-二甲基乙酰胺作为反应溶剂,保持化合物1投料量21.6 g(0.1 mol)不变,n(化合物1) ∶n(氰乙酸乙酯)=1.0 ∶1.3,反应温度60~70 ℃,反应时间3 h,考察了碱的种类和用量对化合物2反应收率的影响,试验数据见图2。
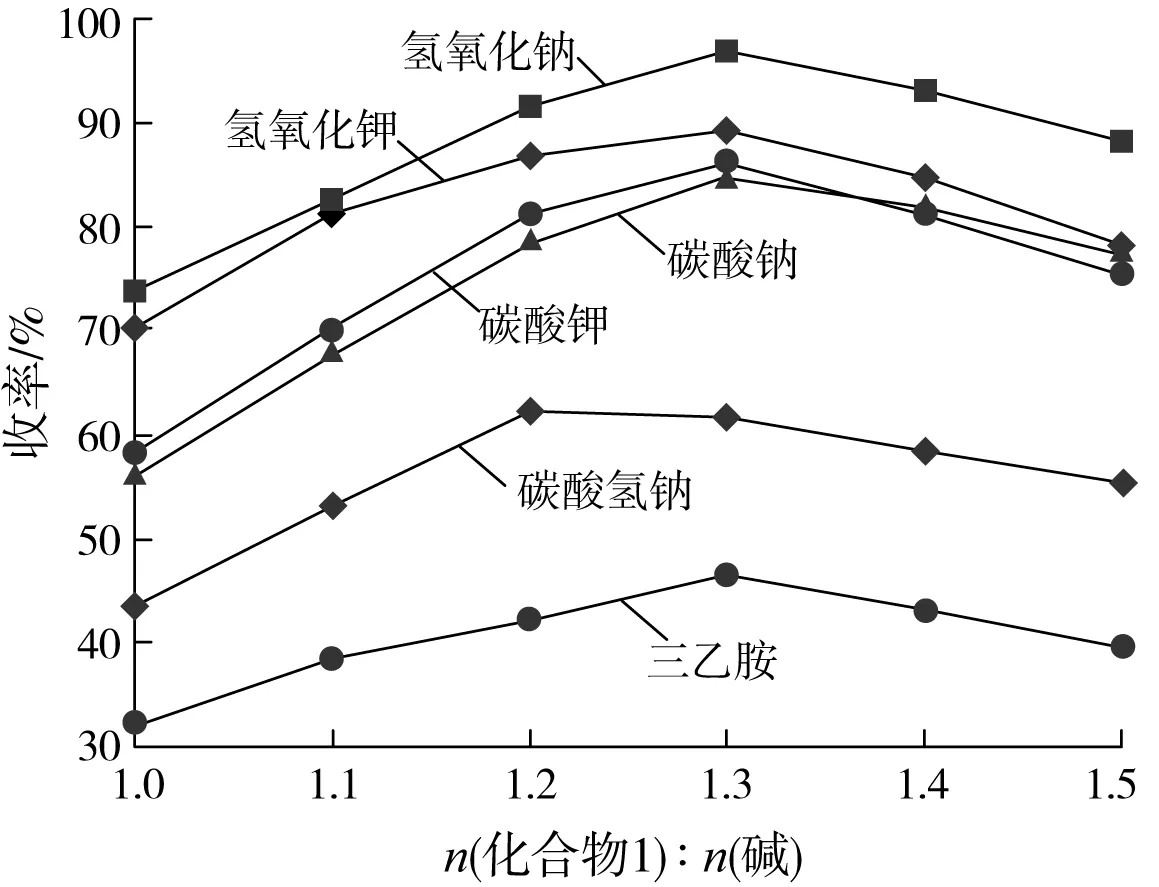
图2 碱的种类和用量对化合物2反应收率的影响
由图2可见,不同强度的碱及碱的用量对反应收率都产生了一定的影响,NaOH作为反应的碱收率最高,KOH由于碱性太强,导致化合物1分子中的氯发生水解,生成水解副产物,致使化合物2的收率降低。而碱性太弱的碱不能为亲核取代提供足够的碱性环境,因而收率也较低。因此,该反应选用NaOH作为反应的碱。
2.1.2溶剂对化合物2收率的影响
保持化合物1投料量21.6 g(0.1 mol)不变,n(化合物1) ∶n(氰乙酸乙酯)=1.0 ∶1.3,n(化合物1) ∶n(NaOH)=1.0 ∶1.3,反应温度60~70 ℃,反应时间3 h,考察了溶剂对化合物2反应收率的影响,结果见表1。

表1 溶剂对产品含量和收率的影响
亲核取代反应需要在极性有机溶剂中进行,采用不同的有机溶剂,由于溶剂在碱性条件下的稳定性不同,反应过程中生成的杂质不同,反应的转化率也存在差异,从而影响到反应的收率。
由表1可知,在所选溶剂和试验条件下,由于N,N-二甲基甲酰胺在反应过程中易生成2-氯-N,N-二甲基-4-三氟甲基苯胺副产物,从而造成反应收率降低,而1,3-二甲基-2-咪唑啉酮和N-甲基吡咯烷酮在碱性条件下稳定性较差,因而,采用这3个溶剂时反应收率和产品含量均偏低,故确定采用N,N-二甲基乙酰胺作为反应溶剂。
2.1.3氰乙酸乙酯用量对化合物2反应收率的影响
以N,N-二甲基乙酰胺作为反应溶剂,保持化合物1投料量14.7 g(0.1 mol)不变,反应温度60~70 ℃,反应时间3 h,考察原料配比对化合物2反应收率的影响,结果见表2。
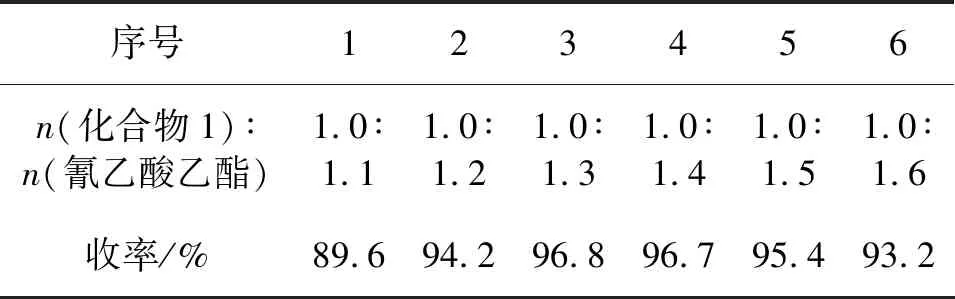
表2 化合物1与氰乙酸乙酯物质的量比
由表2可见,随着化合物1与氰乙酸乙酯物质的量比增大,转化率逐渐上升,这可能是因为随着两者物质的量比增大,反应体系原料浓度增加,导致反应转化率上升。随着两者物质的量比增大,化合物2的收率呈峰值变化趋势,当两者物质的量比为1.0 ∶1.3时,收率最高,为96.8%,当两者物质的量比继续增大时,副反应增加,收率反而降低,因此,选择两者物质的量比为1.0 ∶1.3。
2.1.4 pH对化合物3收率的影响
以N,N-二甲基乙酰胺作为反应溶剂,保持化合物2投料量14.7 g(0.05 mol)不变,在110~120 ℃滴加浓硫酸,反应时间3 h,考察了反应体系pH对化合物3收率的影响,结果见表3。
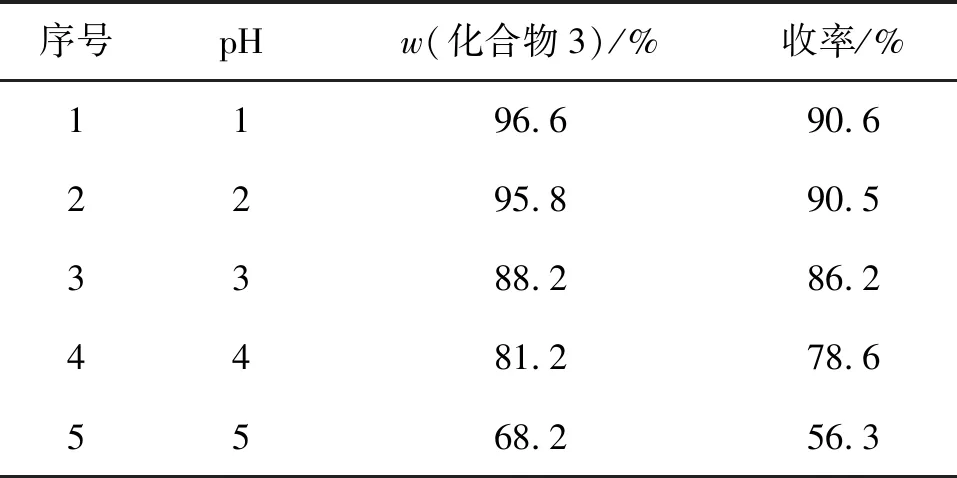
表3 反应体系pH对产品含量和收率的影响
合成化合物3的反应是一个脱羧反应,在酸性条件下,质子和酯羰基的氧结合增加了羰基碳的亲电性,更加有利于水分子的进攻,从而使原料脱羧得到产品。
由表3可知,当反应体系pH为4~5时,产品含量和收率均较低,随着pH下降,收率逐渐提高,pH为1~2时,反应收率大于90%。故确定脱羧反应体系的最佳pH为1~2。
2.1.5脱羧反应温度对化合物3收率的影响
以N,N-二甲基乙酰胺作为反应溶剂,保持化合物2投料量14.7 g(0.05 mol)不变,控制反应体系的pH为1~2,滴加浓硫酸,反应时间3 h,考察了反应温度对化合物3收率的影响,结果见表4。

表4 反应温度对产品含量和收率的影响
脱羧反应温度主要影响反应的快慢和杂质的多少。在合适的反应温度下,反应速率大,杂质少,收率高。由表4可知,脱羧反应收率、产品含量随温度升高而升高,在110~120 ℃达到最高点,温度继续升高则呈下降趋势,原因可能是温度太高,副反应速率增大,导致反应收率下降。
合成化合物3的最佳工艺条件为:亲核取代反应以N,N-二甲基乙酰胺作为反应溶剂,化合物1与氰乙酸乙酯、氢氧化钠物质的量比为1.0 ∶1.3 ∶1.3,反应温度60~70 ℃,反应时间3 h;脱羧反应在第一步反应体系中进行,在110~120 ℃滴加浓硫酸,控制体系的pH为1~2。在此工艺条件下,化合物3的收率可达88.0%。
2.2 化合物4的合成工艺条件优化
还原步骤的主要副产物为偶联副产物双[2-(3-氯-5-三氟甲基吡啶-2-基)乙基]胺和脱氯副产物N-{2-[5-(三氟甲基)-吡啶-2-基]乙基}乙酰胺以及焦油。主要考察了反应温度对反应收率的影响。
保持化合物3投料量11.3 g(0.05 mol)不变,钯碳与化合物3的质量比为1.000 ∶0.015、氢气压力为3.0 MPa,考察反应温度对杂质含量和化合物4收率的影响,结果见表6。
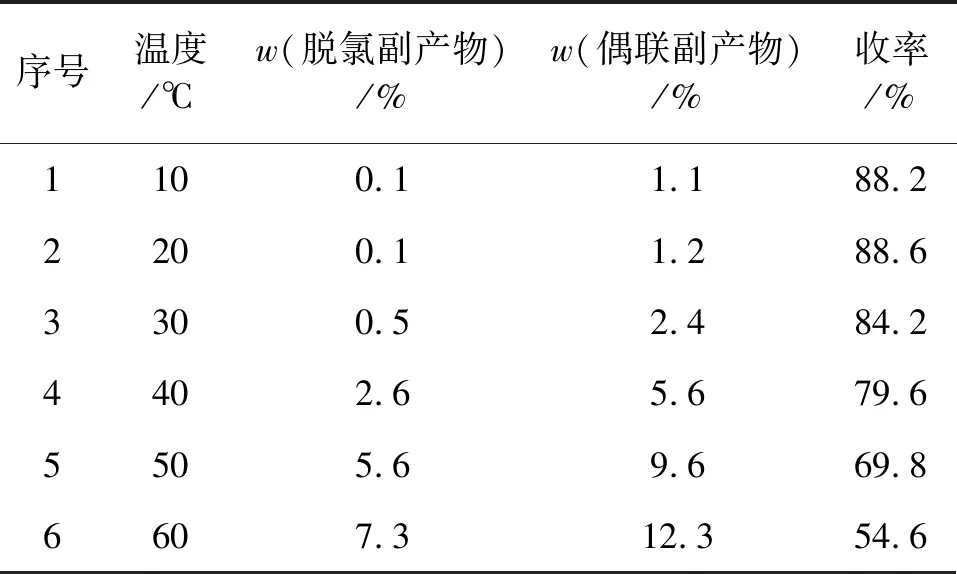
表6 反应温度对产品含量和收率的影响
由表6可见,当反应温度为20 ℃ 时,收率最好,为88.6%;当温度升至60 ℃时,收率只有54.6%。这是因为随着反应温度的升高,偶联副产物和脱氯副产物的含量均增加,并且焦油含量也随之增加。故最佳的反应温度为20 ℃。
2.3 化合物5的合成工艺条件优化
酰胺的水解可以在酸性条件下进行,也可以在碱性条件下进行,但由于吡啶环上含有氯原子,在碱性条件下会产生脱氯杂质,故水解反应不能在碱性条件下进行。并且由于吡啶环上含有三氟甲基,在硫酸水解的条件下,可能会产生少量三氟甲基水解的产物,因此,该步水解采用盐酸。
在盐酸催化下,酰胺分子中的羰基氧原子质子化形成中间体Ⅰ,质子化的羰基碳的亲电性大大增强,更容易受亲核能力不强的水分子进攻,形成四面体中间体Ⅱ,再经过质子转移形成中间体Ⅲ,最后消去质子得到Ⅳ和乙酸,Ⅳ与盐酸成盐得到Ⅵ,使反应平衡向右移动,最终使反应完全。可能的水解反应机理如图3所示。
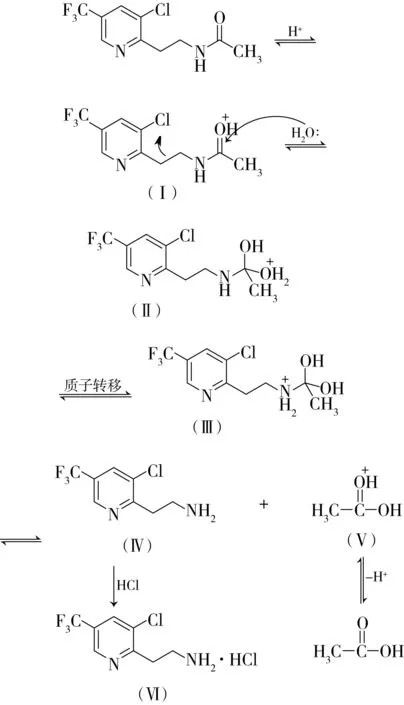
图3 酰胺水解反应机理
酰胺水解反应主要受盐酸用量和水解反应温度的影响,因此,考察了盐酸用量和反应温度对反应收率的影响。
2.3.1盐酸用量对化合物5收率的影响
保持化合物4投料量13.4 g(0.1 mol)不变,反应温度90 ℃、反应时间8 h,考察盐酸用量对反应收率的影响,结果见表7。
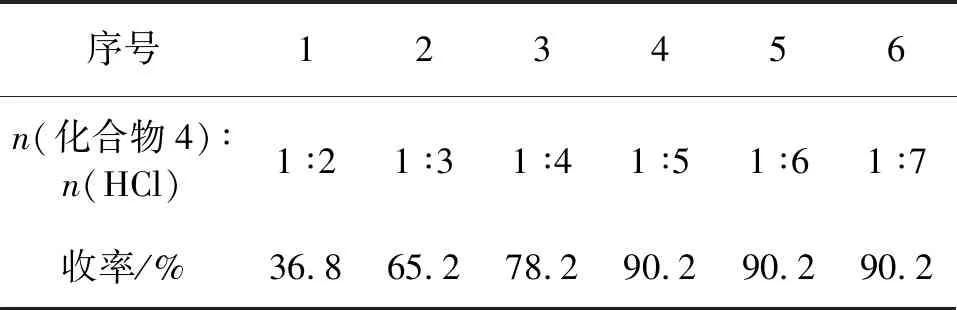
表7 盐酸用量对化合物5收率的影响
由表7可知,当化合物4与盐酸物质的量比为1 ∶5时,收率最高,为90.2%。在此基础上继续增加盐酸的量并没有显著提高反应的收率。
2.3.2反应温度对收率影响
保持化合物4投料量13.4 g(0.1 mol)不变,化合物4与盐酸物质的量比为1 ∶5、反应时间8 h,考察反应温度对反应收率的影响,结果见表8。
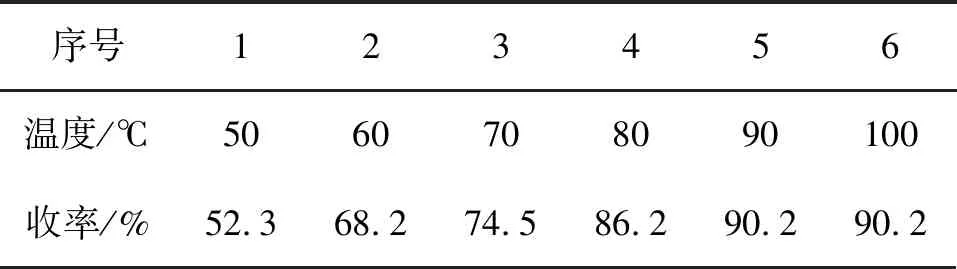
序号123456温度/℃5060708090100收率/%52.368.274.586.290.290.2
由表8可知,反应收率随着反应温度的升高而增加。这是因为酰胺的水解反应比其他羧酸衍生物的水解反应需要更高的温度,酰胺水解需在强酸和接近回流温度的条件下进行。该反应的最佳反应温度为90 ℃ 。
综上所述,合成化合物5的最佳反应条件为:化合物4与盐酸的物质的量比为1 ∶5,90 ℃反应8 h,收率90.2%。
3 结论
研究了3-氯-5-(三氟甲基)-2-乙胺基吡啶的合成工艺并进行了优化。以低成本的2,3-二氯-5-三氟甲基吡啶为起始原料,经亲核取代和脱羧“一锅法”以88.0%的收率合成了化合物3,比文献[10]报道的收率高7%,并实现了溶剂DMAC的回收和重复利用;研究了反应温度对化合物3还原反应偶联杂质和脱氯杂质的影响,确定了适宜的还原反应温度为20 ℃;探讨了盐酸用量和反应温度对水解反应收率的影响,确定了化合物4与氯化氢物质的量比为1 ∶5,反应温度为90 ℃。
优化后的合成工艺具有反应条件温和、操作简单、反应总收率高和环境友好等优点,可以直接应用于工业化生产。
猜你喜欢
世界农药(2022年10期)2022-11-10
浙江化工(2022年8期)2022-09-05
当代化工研究(2022年11期)2022-06-27
安徽化工(2022年1期)2022-02-15
能源化工(2021年2期)2021-12-30
中华养生保健(2020年3期)2020-11-16
有机氟工业(2020年2期)2020-07-04
化工学报(2020年4期)2020-05-28
今日农业(2019年11期)2019-08-13
浙江化工(2015年4期)2015-11-28
