
编码Ag85A基因突变体的设计与合成
2019-05-16 06:35赵乐恒吕昌龙翟景波
微生物学杂志 2019年2期
赵乐恒, 翟 郑, 吕昌龙,3, 翟景波,3*
(1.内蒙古民族大学 布鲁氏菌病研究所,内蒙古 通辽 028000; 2.内蒙古自治区布鲁氏菌病防治工程技术研究中心,内蒙古 通辽 028000; 3.中国医科大学 免疫学教研室,辽宁 沈阳 110122)
自Wolff等[1-2]发现裸DNA经肌肉注射可在肌细胞内表达并引入DNA疫苗概念以来,人们对DNA疫苗进行了广泛研究,取得了令人瞩目的进展[3-6]。在以往广泛进行的DNA疫苗构建、接种后免疫应答(固有免疫和适应性免疫应答)或耐受,以及效应产物作用机制研究基础上,虽然DNA疫苗已用于某些动物疾病的治疗,但在人类尚缺乏有效性应用[7-9]。限制DNA疫苗在临床应用的主要问题是低免疫原性,接种后不能获得强有力的保护性免疫应答。在本研究组完成国家自然科学基金资助项目“自制口服DNA疫苗的黏膜表达部位与诱导肠IEL效应机制的研究”的研究过程中,发现应用编码Ag85A全基因制备的可表达载体转化的抗原提呈细胞(DC2.4),在Ag85A可检出mRNA,未检出蛋白分子表达的情况下,能诱导DC2.4表面活化分子的表达及适应性免疫应达的产生,提示可能存在DNA或RNA水平转导抗原信息的可能性及其信号转导途径[10]。为进一步探讨新的抗原提呈机制(RNA Sensors and Adaptors)[11-12],单一使用既转录mRNA又翻译Ag85A蛋白分子的Ag85A DNA质粒疫苗,尚不能解决疫苗DNA是否存在或能否通过RNA或DNA传感器从抗原提呈细胞(DC2.4)直接将抗原信息提呈给Th细胞,诱导适应性免疫应答可能机制。为此,本研究自行设计了只转录mRNA活性,不翻译Ag85A抗原蛋白分子的Ag85A突变体(Ag85A-M)序列,利用普通PCR法,进行全基因合成,并构建突变体pIRES2-EGFP-Ag85A-M载体。同时构建既具有转录mRNA活性又能翻译成抗原蛋白的pIRES2-EGFP-Ag85A载体作为对照,用于后续细胞内抗原信息核酸水平转导提呈机制的研究。
1 材料与方法
1.1 材料
1.1.1 菌株与质粒 菌株E.coliDH5 Code No.9057,购自大连Takara公司;质粒pMDTM19-T Vector Coke No.6023,购自大连宝生物公司;pIRES2-EGFP购自BD Biosciences Clontech;细胞DC2.4来自中国医科大学免疫学教研室。
1.1.2 试剂TaqDNA聚合酶、PrimeSTARTMHS DNA Polymerase、10 mmol/L dNTPs Mix、DL2000 Marker、限制性内切酶、DNA纯化试剂盒和琼脂糖 (Agarose)购自大连宝生物公司;寡核苷酸 (Oligonucleotide)购自上海生工;胰化蛋白胨(Bacto-tryptone)和酵母提取物(Bacto-yeast extract)购自Oxiod公司;其他化学试剂购自Sigma或Aldrich公司。
1.2 方法
1.2.1 分子生物学方法 本研究中所用方法均为分子生物学标准操作方法[13]。Real-time PCR,提取瞬转细胞系DC-Ag85A和DC-Ag85A-M总RNA,取10 μL RNA进行逆转录成cDNA,采用 β-actin作为内参进行相对定量分析。定量反应体系按照12.5 μL SYBR Premix ExTaq,1 μL PCR Forward Primer, 1 μL Uni-miR qPCR Primer,2 μL cDNA模板,8.5 μL ddH2O,总体积为25 μL,每个样品设置3个平行孔。扩增程序:预变性95 ℃ 30 s;95 ℃ 5 s,60 ℃ 20 s,40个循环;采用2-ΔΔCt法分别计算Ag85A(M) mRNA的相对表达量;Western blot检测Ag85A蛋白表达,提取瞬转细胞系DC-Ag85A和DC-Ag85A-M总蛋白,BCA法测定蛋白浓度,沸水变性,配制10%的分离胶及5%的浓缩胶,SDS-PAGE电泳。电泳结束后,根据分离胶面积,切割同样大小的6张滤纸及1张PVDF膜。将PVDF膜用甲醇活化10 s后,同滤纸、分离胶放入转移缓冲液中浸泡10 min。将分离胶、PVDF膜及滤纸按照“三明治”顺序(负极-滤纸-分离胶-PVDF膜-3层滤纸-正极)铺好,并小心赶走滤纸和分离胶之间的气泡。按照1 mA/cm2电流量设置恒定电流为48 mA,半干式电转法转膜40 min。转膜结束后,取出PVDF膜,观察预染Marker的转移情况,并据此判断目的蛋白的转移情况。将封闭液浸没PVDF膜,封闭后室温摇床反应1 h,并用TBS轻微洗涤数次。加入用封闭液稀释的相应一抗anti-Ag85A,室温摇床孵育2 h,并用TBS洗膜3次,每次15 min。再加入用封闭液稀释的相应二抗室温摇床孵育1 h,并用TBS洗膜3次,每次15 min。ECL显色检测。
1.2.2 编码Ag85A基因突变体的设计 编码Ag85A基因全长891 bp(GenBank Accession: CP003248.1)。以其起始密码子ATG中碱基A为1计算,将114位和230位的C替换为G,使含有114位和230位碱基的密码子称为终止密码,此为编码Ag85A基因突变体(Ag85A-M)。
1.2.3 引物设计 ①拆分小片段合成法的引物设计方案:为正确合成编码Ag85A-M全基因片段,采用分段PCR合成法, 即将基因拆分成3个小片段(A:332 bp,B:332 bp,C:267 bp)和2个小片段(D:488 bp,E:423 bp)进行分段合成。3小片段拆分合成的引物设计方案见图1,2小片段拆分合成的引物设计方案见图2。除C1F、C1R为64个碱基,C3R为62个碱基外,其余引物长均为59个碱基。 引物间重叠(overlapping)20个碱基,F引物取正向序列,R引物取反向互补序列。以同样方案合成正常Ag85A全基因片段作为对照。②引物合成:按上述引物设计序列合成所需 PCR引物,引物由上海生工公司合成。引物序列见表1 和表2。

图1 Ag85A-M三小片段拆分合成的引物设计方案Fig.1 3 Ag85A-M Segment splitting primer design strategy

图2 Ag85A-M二小片段拆分合成的引物设计方案Fig.2 2 Ag85A-M Segment splitting primer design strategy
1.2.4 全基因合成技术路线 Ag85A-M和Ag85A 拆分小片段和最终全基因的合成路线: 全长基因小片段拆分→引物设计、合成→分段延伸→单段PCR扩增→切胶回收→单段插入片段制作→单段克隆→转化、检菌、质粒提取、测序→PCR扩增单段基因→切胶回收→PCR扩增全长基因→切胶回收→加A→全长基因克隆、测序、双酶切验证。
1.2.5 PCR分段延伸 PCR反应体系组成:Primer F方向(20 pmol/μL)各加入2 μL,Primer R方向(20 pmol/μL)各加入2 μL,dNTP Mixture(各2.5 mmol/L)加入8 μL,5×PrimeSTARTMBuffer加入20 μL,PrimeSTARTMHS DNA Polymerase(2.5 U/μL)加入1 μL,用双蒸水补充至100 μL。PCR反应条件:94 ℃ 5 min,缓慢降温10 min至55 ℃,55 ℃ 5 min,72 ℃ 5 min,4 ℃保存。

表1 拆分成3个小片段PCR合成设计应用的引物序列

表2 拆分成2个小片段PCR合成设计应用的引物序列
1.2.6 单段PCR扩增 ①正常GC含量片段扩增:PCR反应体系组成为Template(延伸产物)加入50 ng,Primer F(5′端)(20 pmol/μL)加入1 μL,Primer R(3′端)(20 pmol/μL)加入1 μL,dNTP Mixture(各2.5 mmol/L)加入8 μL,5×PrimeSTARTMBuffer加入20 μL,PrimeSTARTMHS DNA Polymerase(2.5 U/μL)加入1 μL,用双蒸水补充至100 μL。PCR反应条件:98 ℃ 10 s,55 ℃ 10 s,72 ℃ 适时,30个循环,4 ℃保存。②高GC含量片段扩增:PCR反应体系组成为Template(延伸产物)加入50 ng,Primer F(5′端)(20 pmol/μL)加入1 μL,Primer R(3′端)(20 pmol/μL)加入1 μL,dNTP Mixture(各2.5 mmol/L)加入16 μL,2×GC BufferⅠ加入50 μL,TaKaRa LATaq(5 U/μL)加入1 μL,用双蒸水补充至100 μL。PCR反应条件:94 ℃ 30 s,55 ℃ 1 min,5个循环;94 ℃ 30 s,55 ℃ 30 s,72 ℃ 适时,30个循环;4 ℃保存。
1.2.7 单段插入(Insert)片段制备 PCR反应体系组成:DNA(PCR回收产物)1 μg,dNTP Mixture(各2.5 mmol/L)加入4 μL,10×PCR Buffer加入5 μL,TaKaRaTaq(5 U/μL)加入0.5 μL,用双蒸水补充至50 μL。PCR反应条件:72 ℃ 20 min,4 ℃保存。
1.2.8 单段克隆 PCR反应体系组成:Insert DNA 100 ng,Vector DNA 50 ng,SolutionⅠ5~10 μL,用双蒸水补充至10~20 μL。反应条件:16 ℃ 60 min。
1.2.9 PCR扩增单段基因 PCR反应体系组成:Template(单段OK质粒)加入10 ng,Primer F方向(20 pmol/μL)加入1 μL,Primer R方向(20 pmol/μL)加入1 μL,dNTP Mixture(各2.5 mmol/L)加入8 μL,5×PrimeSTARTMBuffer加入20 μL,PrimeSTARTMHS DNA Polymerase(2.5 U/μL)加入1 μL,用双蒸水补充至100 μL。PCR反应条件:98 ℃ 10 s,55 ℃ 10 s,72 ℃ 适时,30个循环,4 ℃保存。
1.2.10 PCR扩增全长基因 PCR反应体系组成:Template(单段)各加入20 ng,Primer F(5′)(20 pmol/μL)加入1 μL,Primer R(3′)(20 pmol/μL)加入1 μL,dNTP Mixture(各2.5 mmol/L)加入8 μL,5×PrimeSTARTMBuffer加入20 μL,PrimeSTARTMHS DNA Polymerase(2.5 U/μL)加入1 μL,用双蒸水补充至100 μL。PCR反应条件:98 ℃ 10 s,55 ℃ 10 s,72 ℃ 适时,30个循环,4 ℃保存。
1.2.11 全长片段填加碱基腺嘌呤(A) PCR反应体系组成:DNA(PCR回收产物)1 μg,dNTP Mixture(各2.5 mmol/L)加入4 μL,10×PCR Buffer加入5 μL,TaKaRaTaq(5 U/μL)加入0.5 μL,用双蒸水补充至50 μL。PCR反应条件:72 ℃ 20 min,4 ℃保存。
1.2.12 全长克隆测序、双酶切 PCR反应体系组成:Insert DNA 各100 ng,Vector DNA 50 ng,SolutionⅠ5~10 μL,用双蒸水补充至10~20 μL。反应条件:16 ℃ 60 min。质粒转化、涂平板、扩培养、质粒提取、纯化,委托上海生工生物工程有限公司测序,双酶切条件按限制性内切酶说明书进行。
1.2.13 瞬转DC2.4细胞 将构建好的质粒用Neon电转仪电转到DC2.4细胞中。电转参数为1 200 V,20 ms,2 pulse。培养条件:McCoy′s 5A,10% FBS,0.5 μg/mL Puromycin,37 ℃,5% CO2。培养时间48 h,胰酶浓度0.05%,胰酶消化时间37 ℃,1 min 20 s。进行基因转录产物mRNA荧光定量PCR检测及翻译产物Ag85A蛋白Western blot检测,引物序列:Ag85A(M)Sense primer 5′-AAGTGGGAGACCTTCCTGACC-3′,Antisense primer 5′-GAAGAAGCAGCCATCGAAAGA-3′;内参β-actin Sense primer 5′-TTGAACATGGCATTGTTACCAA-3′,Antisense primer 5′-TGGCATAGAGGTC-TTTACGGA-3′[14]。
1.2.14 统计学结果分析 采用GraphPad Prism 5 One-way ANOVA(and nonparametric)进行柱形图分析;采用SPSS 19 单样本T检验进行单柱3样本Sig分析。
2 结果与分析
2.1 A、B、C、D和E片段合成
采用普通PCR法分段扩增合成的A、B、C、D和E小片段均获得预期结果,即A和B小片段为332 bp, C为264 bp, D为488 bp,E为423 bp。结果见图3 。
2.2 双酶切检测
应用限制性内切酶EcoR I/BamH I双酶切pIRES2-EGFP-Ag85A-M重组载体进行检测鉴定,获得了Ag85A-M 891 bp的全基因片段条带,和5.3 kpb的pIRES2-EGFP 载体片段条带。条带大小正确,见图4。


图3 PCR法合成的片段A、B、C、D和E 琼脂糖凝胶电泳结果Fig.3 Agarose gel electrophoresis of synthesized segment A,B,C,D and E by PCR 1.5%gel,1×TAE;M:Marker DL2000

图4 双酶切重组载体的琼脂糖凝胶电泳结果Fig.4 Agarose gel electrophoresis pattern ofEcoR I/BamH I for digestion of pIRES2-EGFP-Ag85A-M carrierA:双酶切pIRES2-EGFP-Ag85A-M;B:双酶切pIRES2-EGFP-Ag85AA:EcoR I/BamH I digested pIRES2-EGFP-Ag85A-M;B:EcoR I/BamH I digested pIRES2-EGFP-Ag85A1% gel, 1×TAE; M: Marker SM0331(Songon)
2.3 全基因片段测序结果
Ag85A-M和Ag85A全基因片段测序结果核苷酸残基构成和排序正确,结果见图5、6。Ag85A-M序列中与预先设计的突变位置及碱基组成一致,结果见图7。
2.4 Ag85A(M)基因转录和Ag85A蛋白表达
从图8和图9可以看出,Ag85A基因既转录mRNA,又表达Ag85A蛋白;突变体Ag85A-M基因只转录mRNA,不表达Ag85A蛋白,与设计一致。由图8可知,定量PCR相对定量Ag85A及Ag85A-M mRNA Fold change值。NC为负对照,值为1.00±0.29(*P<0.05,n=3);DC-Ag85A,14.35±0.96(**P<0.01,n=3);DC-Ag85A-M,15.78±0.83(***P<0.001,n=3)。由图9可知,Ag85A及Ag85A-M Western blot电泳,泳道1,NC(负对照),无目标带;泳道2,DC-Ag85A,目标带明显;泳道3,DC-Ag85A-M,无目标蛋白带。对照为内参HPRT蛋白,表达量多。

图5 pIRES2-EGFP-Ag85A-M载体的Ag85A-M序列分析Fig.5 Ag85A-M Sequencing of pIRES2-EGFP-Ag85A-M carrier

图6 pIRES2-EGFP-Ag85A载体的Ag85A序列分析Fig.6 Ag85A Sequencing of pIRES2-EGFP-Ag85A carrier
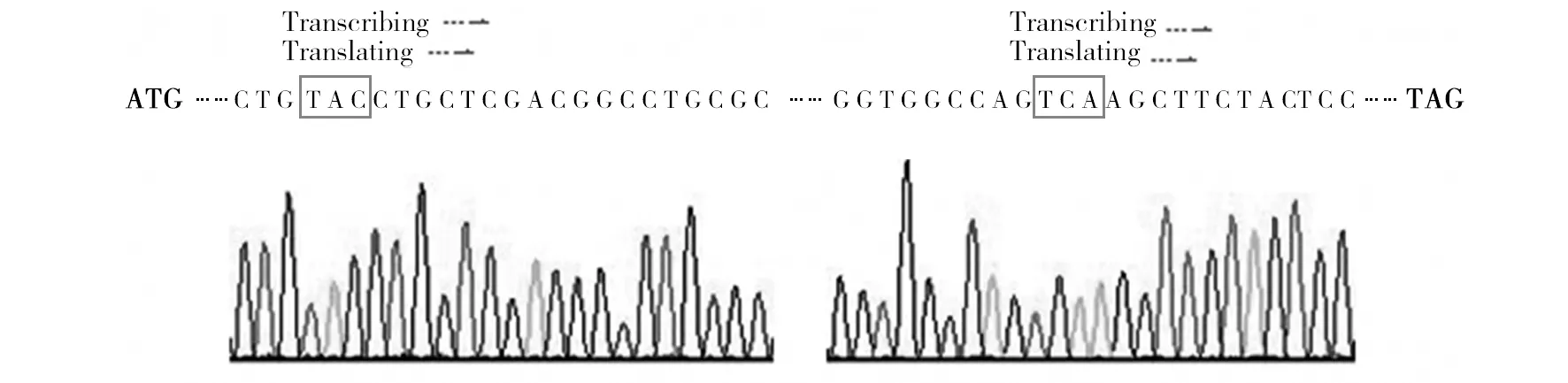

图7 Ag85A-M 和 Ag85A对应部位的序列比较

图8 DC2.4细胞中Ag85A及Ag85A-M基因的mRNA表达Fig.8 The expression of Ag85A and Ag85A-M mRNA in DC2.4
2.5 三片段拆分法与二片段拆分法合成效果
从A、B、C、D和E小片段克隆形成的菌落中分别随机挑取8个菌落进行质粒提取和测序,A、B、C阳性克隆率平均值为87.67%,基因保真度为90.33%;D、E阳性克隆率平均值为81.5%,基因保真度为23%。两种基因片段拆分方法人工合成片段的阳性克隆率差别不大(P>0.05),但332 bp及以下片段PCR法合成保真度显著高于423 bp及以上片段(P<0.000 1)。见表3和图10。

图9 DC2.4细胞中Ag85A及 Ag85A-M基因的蛋白表达Fig.9 The expression of Ag85A and Ag85A-M protein in DC2.4
1:DC2.4细胞(NC);2:含有Ag85A范围的DC2.4细胞;3:含有Ag85A-M基因的DC2.4细胞
1:DC2.4(Negative Contrast);2:DC2.4 transformed with Ag85A;3:DC2.4 transformed with Ag85A-M

表3 三片段拆分法与二片段拆分法合成效果分析

图10 三分法与二分法合成片段的阳性率和准确率比较Fig.10 Positive rate and Fidelity rate contrast of 3 Splitting and 2 Splitting***P<0.000 1
3 讨 论
目前获得目的基因的方法主要有两种:一是采用PCR的方法对模板的目的区域直接进行扩增;二是利用DNA合成仪器直接合成单链的DNA片段。前者要求事先具备高质量的DNA模板,后者在保证合成质量的前提下,合成的DNA 片段长度一般不超过100 bp。若想获得长度大于100 bp的基因,在没有合适的 DNA模板的情况下,以上两种方法显然都不适用。随着全基因合成技术的发展,不依赖模板的功能性蛋白质、多肽全基因合成的需求量会越来越大,全基因合成技术必将广泛应用。基于PCR 技术的基因合成方法的主要工作是引物的设计和合成,按Stemmer 的理念,每条引物长40 bp,相邻引物互补20 bp;设计的引物与做定点突变设计引物一样,其设计空间比较小。目前在进行DNA片段拆分时,倾向于改变引物的长度,在线设计引物软件Gene2Oligo网址为http://berry.engin.umich.edu/gene2oligo/。近来较新的报道合成基因的方法大多是在Stemmer的设计理念基础上做改变,如将overlap extension PCR(OE-PCR)与dual asymmetrical PCR (DA-PCR)联合使用等[15]。国际权威实验技术专著 《PCR primers:A laboratory manual》[13]也应用了Stemmer的方法,虽然在具体步骤方面做了一定的修饰,但效果几乎一样,时间得到缩短。 目前每天化学合成的寡核苷酸数以十万计,在人类基因组计划(HGP)、基因治疗、基因芯片等生物医学热点中有着广泛应用。DNA的人工合成正在分子生物学和医学等许多领域中发挥越来越多的作用。
目前困扰全基因合成技术的问题主要有两个:一是合成基因中碱基错误率高,二是合成成本高。本研究从这两方面出发,对基因合成的全基因拆分方案进行了探讨。以合成Ag85A全长基因891 bp、重迭片段长度20 nt为例,可以看出拆分的寡核苷酸长度越短,合成的寡核苷酸数量越多,伴随合成的核苷酸总数也越多,总费用也随之增加。由于DNA合成公司在59 nt和60 nt寡核苷酸之间的收费有断档,所以当寡核苷酸拆分长度由59 nt增加到60 nt,合成总费用将剧增。本研究以在基因合成过程中降低合成基因中的错误率,降低合成过程的工作量,降低基因合成的成本为目标,将Ag85A全长基因进行适当大小片段拆分设计,采用三片段法和两片段法拆分进行了探讨,获得与设计一致的结果。两种基因片段拆分方法虽然各片段间阳性克隆率差别不大,但332 bp及以下片段PCR法合成保真度显著高于423 bp及以上片段(P<0.000 1)。该结果提示对于基因长度在891 bp左右的片段进行三小片段拆分,或者拆分片段成264~332 bp大小进行PCR合成所得的目的基因片段更好。
本研究通过三小片段拆分,经PCR扩增的定点突变的Ag85A-M和正常Ag85A全基因片段分别进一步克隆到pIRES2-EGFP真核表达载体,瞬转至DC2.4细胞中获得了阳性表达,即Ag85A-M转录 mRNA,不表达相应蛋白分子;作为对照的Ag85A则即转录mRNA,也表达蛋白分子。并在Ag85A基因转化的DC2.4细胞内证明了RNA传感器的存在[14]。
猜你喜欢
中国人兽共患病学报(2022年9期)2022-10-19
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
中国农学通报(2022年12期)2022-06-01
中国糖料(2022年2期)2022-04-06
中国种业(2021年11期)2021-11-25
科学导报(2021年29期)2021-06-03
教学考试(高考生物)(2021年2期)2021-05-31
江西农业学报(2021年4期)2021-04-20
中国生殖健康(2020年4期)2021-01-18
