
MicroRNAs contribute to ATP-binding cassette transporter- and autophagy-mediated chemoresistance in hepatocellular carcinoma
2019-05-17 02:29MarEspeltMarBacigalupoPabloCarabiasMarTroncoso
World Journal of Hepatology 2019年4期
María V Espelt, María L Bacigalupo, Pablo Carabias, María F Troncoso
Abstract
Key words: Hepatocellular carcinoma; Chemoresistance; ABC transporter; Autophagy;miRNA
INTRODUCTION
Hepatocellular carcinoma (HCC) accounts for around 80%-90% of liver cancer. It's the fifth most common cancer worldwide and the third cancer-related death cause. In general, HCC develops in a background of cirrhosis. The main factors that contribute to this tumor are infections with hepatitis B or C virus, alcohol abuse and nonalcoholic fatty liver disease[1]. Despite the development of early detection methods,80% of HCC patients are diagnosed at a stage of the disease so advanced that the patient's survival is just of about few months[2]. When HCC is diagnosed at early stages the most effective approaches are partial hepatectomy or liver transplantation,however in most patients the tumor is not detected promptly or it is hard to find a compatible donor. In intermediate stages, transarterial chemoembolization is the treatment of choice[3]. For patients with unresectable HCC, sorafenib oral administration slightly improves survival[4]. Even the ones who qualify for surgery have a modest improvement in the survival because of its high rate of recurrence,occurring in more than 90% of patients[2].
Treatment with anti-cancer drugs (chemotherapy) can destroy tumor cells helping patients to control cancer growth. Some of these drugs to treat HCC are 5-fluorouracil(5-FU), cisplatin, doxorubicin[5], paclitaxel and the multitarget tyrosine kinase inhibitor sorafenib. But unfortunately, liver cancer patients usually develop drug resistance to chemotherapy[6]. Cancer drug resistance is a multifactorial phenomenon involving many different mechanisms such as gene mutation, DNA repair pathway aberrations, impairment of the apoptotic machine, alterations in drug metabolism and processing, activation of cell survival signaling and escape of drug sensitivity,autophagy, epigenetic regulation, altered lipid metabolism and tumor microenvironment participation[2]. Among these mechanisms, modifications in drug uptake or efflux produce a diminished intracellular drug concentration, leading to tumor cell survival and resistance to death. Until some years ago, the reduction of intracellular drug concentration was thought to be the only important mechanism for resistance. Nevertheless, many other processes have been appeared to be involved more lately. Autophagy is one of these novel processes that also contribute to tumor chemotherapy resistance; moreover, autophagy inhibitors are used to sensitize different cancers to chemotherapy. Under this scenario, microRNAs (miRNAs) are nowadays emerging as master regulators of normal physiology- and tumor-related gene expression. Thus, it is not surprising that these molecules can also regulate chemoresistance.
In this review we summarize the findings from the last decade regarding the molecular mechanisms of chemoresistance in HCC, focusing on the involvement of miRNAs. We especially highlight the role of these molecules in drug efflux through ABC (ATP-binding cassette) transporters and autophagy.
miRNAs IN THE DEVELOPMENT OF HCC CHEMORESISTANCE
miRNAs are short RNA molecules of 18-25 nucleotides in length. They are non-coding molecules that regulate the expression of many genes. By binding to the 3'-untranslated region (UTR) of target genes through base complementarity, miRNAs lead to mRNA degradation or translational repression, acting as negative regulators of gene expression[6]. Many reports have described a role of these molecules in the control of diverse biological processes such as development, differentiation, cell proliferation and apoptosis[7]. They can also act as oncogenes or tumor suppressors in different human cancers.
Up to date more than 2500 miRNAs have been described in humans and each of them has been reported to silence more than one gene. miRNAs take a crucial part in different processes in normal and abnormal liver development. They are involved in lipid, cholesterol and glucose metabolism, and liver differentiation (see Ref.[8]review). They take part in apoptosis, necrosis, cell cycle, proliferation, epithelialmesenchymal transition and inflammation[9]. Further, studies derived from the comparison between normal liver and human liver cancer showed the aberrant expression of many miRNAs in cancer cells. It was interesting to find that some of them such as, miR-21, miR-221 and miR-222 are upregulated in HCC whereas miR-122, miR-199 and the let7 family members are downregulated[7], showing the complex pattern of miRNA expression in this type of cancer. Regarding the molecular mechanisms, protein kinase B (AKT), mammalian target of rapamycin (mTOR),Wingless-type MMTV integration site family (Wnt), Janus kinase/signal transducers and activators of transcription (JAK/STAT), and Mitogen-activated protein kinases(MAPK) are the main miRNA-regulated pathways during HCC tumorigenesis[10].
Thus, many miRNAs are deregulated in HCC and some of them participate in the progression of chemoresistance[11]. Nevertheless, their precise roles in the development of drug resistance in liver cancer are not fully understood. In the following sections we particularly analyze the relationship between miRNAs and ABC transporter-mediated drug resistance in HCC. Besides, we summarize the novel evidence that relates miRNAs, autophagy and chemoresistance in HCC.
miRNAs AND ABC TRANSPORTER-MEDIATED DRUG RESISTANCE IN HCC
The ABC transporters are cell membrane proteins that couple the hydrolysis of ATP to extrude different types of xenobiotics and metabolites against concentration gradient. Efflux transporters such as P-glycoprotein (P-gp/ABCB1), breast cancer resistance protein (BCRP/ABCG2) and multidrug resistance-associated protein 1(MRP1/ABCC1), among others, limit the exposure to chemotherapeutic drugs by extruding them from cells. Consequently, these transporters are main actors in the drug resistance phenomenon. Furthermore, their overexpression in HCC cells represents a big obstacle for chemotherapy treatment and the central mechanism that contributes to drug resistance[12]. Here we summarize the recent studies describing the role of major miRNAs on ABC transporter family proteins and their impact on HCC chemoresistance.
miR122
This is a liver specific miRNA[13]and it participates in lipid and cholesterol metabolism and its decreased expression has detrimental effects on the liver[8].Frequently, miR122 is downregulated in HCC tumors denoting poor prognosis and metastatic properties[14]. In human HCC cell lines (HepG2, HuH-7, Hep3B), miR122 levels were also found reduced. Interestingly, when these cells were adenovirustransduced to overexpress miR122 they became more sensitive to DOX- and vincristine-induced death. Remarkably, miR122-overexpressing HCC cells showed reduced levels ofABCB1andABCC1mRNA expression (Table 1). The latter was also found downregulated at the protein level[12]. These results indicate that miR122 controls sensitivity to drug-induced HCC cell death by downregulating P-gp and MRP1 expression.
miR27a
miR27a is involved in tumorigenesis in different types of cancers. In leukemia cells it was shown that resistance to DOX correlates with high expression of P-gp and low levels of miR27a. The upregulation of this miRNA produced greater mortality in the presence of DOX than the observed in control cells, demonstrating the role of miR27a in chemoresistance in leukemia cells[15]. On the contrary, miR27a upregulation lead tothe overexpression of P-gp in ovarian cancer and cervical carcinoma cells[16].

Table 1 miRNAs and ABC transporter-mediated drug resistance in hepatocellular carcinoma cells
There was no information about the relationship between the miR27a and ABC transporters in HCC until 2013. Chenet al[17]developed Bel-7402 HCC cells resistant to 5-Fluoroacil (5-Fu). They found that these cells overexpressed P-gp while miR27a was almost undetectable. But when cells were transfected with miR27a, the authors observed more sensitivity to 5-Fu, DOX and mitomycin, and diminished expression of P-gp[17](Table 1). Thus, levels of miRNA27a in HCC cells are low and seem to negatively correlate with P-gp expression.
miR503
This molecule is downregulated in some types of cancers such as gastric and endometrial tumors, while it is overexpressed in others, like retinoblastoma or parathyroid carcinoma[18]. In HCC samples, this miRNA is downregulated with respect to non-tumor liver tissues, and this low level is related to malignant progression[19]. Furthermore, the expression of miR503 in different HCC cell lines is also decreased. Wanget al[20]found that miR503 was reduced in DOX-resistant HepG2 HCC cells. Besides, these cells overexpressed both P-gp and MRP1 transporters[20].Further, HepG2 cells transfected to overexpress miR503 showed a reduction in the expression of both transporters at protein level, and at mRNA level as well (Table 1).Interestingly, cells overexpressing miR503 also showed an increased sensitivity to DOX and accumulated more intracellular rhodamine-123[20]. This fluorophore is known to be extruded from the cells through P-gp, therefore these results indicate that miR503 controls sensitivity to DOX-induced death in HepG2 cells by reducing Pgp and MRP1 expression and increasing intracellular drug concentration.
miR375
Initially, this molecule was identified in pancreatic cells. It regulates insulin secretion so it takes part of glucose homeostasis. In many cancers, it is downregulated and acts as a tumor suppressor[21]. In HCC, miR375 is one of the most downregulated miRNAs in tumor tissues and cell lines. Its ectopic overexpression was demonstrated to decrease liver cancer cell growth and invasion, and to promote apoptosis[21,22].Moreover, miR-375 suppressed astrocyte elevated gene-1 (AEG-1) expression by binding directly to its 3′-UTR[22], and AEG-1 overexpression increased P-gp protein levels in HCC cells[23].
In a recent study, DOX-resistant HepG2 cells were established and they overexpressed P-gp. However, when miR375-containing lipid-coated nanoparticles were delivered to these cells, P-gp expression decreased under control cell levels.Also, in cell viability assays, the half inhibitory concentration (IC50) of different HCC cell lines (HepG2, HuH-7, Hep3B and HepG2 resistant to DOX) incubated with nanoparticles containing both DOX and miR375 was lower than the IC50of cells cultured with DOX or nanoparticles containing DOX alone (Table 1). Importantly,nanoparticles loaded with miR375 suppressed liver tumor growth, enhanced the therapeutic effect of DOX and reduced drug toxicity in mice[24].
miR133a
This miRNA is downregulated in breast cancer cell lines and tissues, and this decrease is related to advanced clinical stages[25]. Besides, it's downregulation has been associated with metastasis and recurrence of prostate cancer[26]. In HCC cell lines and tissues, miR133a was also found downregulated and its low expression was related with poor differentiated tumors[27]. Interestingly, predictions performed with computational programs revealed that the 3'UTR of MRP1/ABCC1transporter gene includes a binding site for miR133a (and also for miR326). By luciferase reporter assay it was confirmed that miR133a binds to the 3'UTR ofABCC1gene and controls MRP1 expression. Remarkably, miR133a overexpression in HepG2 cells induced a significant decrease in MRP1 mRNA and protein levels (Table 1). Besides, in the presence of DOX, cells overexpressing miR133a were more sensitive to drug-induced death than control cells[28].
miR326
This miRNA was found to be downregulated in gastric cancer[29]. In breast cancer cells and tissues its expression negatively correlated with MRP1 transporter levels.Furthermore, elevated miR326 levels sensitized cells to cytotoxic drugs[30]. In human HCC tissues, this miRNA was also found downregulated and its reduced expression correlated with tumor malignancy and patient lymph node metastasis.In vitro,miR326 inhibited tumor cell proliferation and invasion, and promoted apoptosisin vivo[31]. Regarding its role as modulator of ABC transporter expression, computational programs predicted a binding site for miR326 at the 3'UTR ofABCC1gene. This binding site was also confirmed by luciferase reporter assay (Table 1). In addition, it was demonstrated that HCC cells overexpressing miR326 had reduced levels of MRP1, both at mRNA and protein levels. Further, miR326-overexpressing HepG2 cells showed more sensitivity to DOX than control cells[28].
miR223
In colon cancer, miR223 is upregulated and acts as an oncogene. On the contrary, it is frequently downregulated in leukemia and lymphomas[32]. In HCC, this miRNA is also reduced and it was shown to induce HepG2 and Bel-7402 cell growth suppression and to promote apoptosis[33]. Moreover, lower miR223 levels were found in HCC patient sera respect to healthy volunteers, and a reduced expression was also determined in liver biopsies compared with normal liver. Thus, this miRNA has been proposed as a novel potential biomarker for HCC[34].
Yanget al[35]showed that P-gp protein expression was higher in some HCC cell lines such as Hep3B, HCC3, LM-6, SMMC7721, HuH-7 and HepG2, than in others like Hep3B and BEL-7402 cells. Moreover P-gp levels positively correlated with DOX IC50.Interestingly, using bioinformatics techniques miR223 was first predicted to bind to the 3'UTR ofABCB1gene. This was later confirmed by performing an EGFP reporter assay where miR223 was proved to specifically interact with the 3'UTR ofABCB1gene. Further, miR223 expression was found to be differentially expressed and inversely correlated with P-gp levels when comparison was made among the eight cell lines. Besides, inhibition of miR223 expression was verified to induce P-gp expression in most of the HCC cell lines analyzed[35]. In addition, transfection of HCC cells with miR223 mimics showed the inhibition of P-gp expression (Table 1), both at mRNA and protein levels (Table 1), and an increase in cell mortality under paclitaxel and DOX treatment. These results were confirmed by upregulating P-gp expression and through a rescue experiment by overexpressingABCB1lacking the 3'UTR.Therefore, this study demonstrates that miR223 has a relevant role in HCC cell chemoresistance by controlling P-gp expression[35].
miR491-3p
In multidrug-resistant cancer tongue cells, this miRNA was found to be decreased and the induction of its expression sensitized cells to chemotherapy[36]. Also in osteosarcoma cells and tissues, miR491-3p was observed downregulated and when its expression was restored, the suppression of tumor growth and invasion was demonstrated[37].
Recently, it was demonstrated that this miRNA is also downregulated in HCC cell lines. Furthermore, in liver tumors miR491-3p levels were reduced compared with matched non-tumor tissues. Besides, P-gp levels were higher in HCC Hep3B, BEL-7402 and SMMC-7721 cells than in normal human liver cell lines (THLE-2 and THLE-3). Notably, P-gp expression inversely correlated with miR491-3p in both tumor and normal liver cells (Table 1). This negative correlation was also found in clinical samples[38]. Moreover, using bioinformatic algorithms, different miRNAs -including miR491-3p- were predicted as candidates to bind to the 3'UTR ofABCB1gene.miR491-3p specific interaction with the 3'UTRs ofABCB1was confirmed performing luciferase reporter gene assays. Interestingly, specificity protein 3 (SP3, a transcription factor suggested to induceABCB1expression) was demonstrated to be another target of miR491-3p. miR491-3p levels inversely correlated with SP3 expression in both HCC cells and liver tumors. Remarkably, Hep3B cells transfected with miR491-3p mimics were more sensitive to DOX and vinblastine. On the contrary, overexpression of P-gp or SP3 restored chemoresistance reverting the sensitivity conferred by miR491-3p.Therefore, a loop including miR491-3p, SP3 and MDR1 in HCC cell chemoresistance was proposed[38].
miR183
The expression of this miRNA is deregulated in some malignancies such as leukemia,breast cancer and liver tumors[39]. miR183 downregulation correlated with metastasis in lung cancer[40]; however, this mRNA was upregulated in HCC tissues as compared to the adjacent non-tumor liver zone[41]. Further, the programmed cell death 4(PDCD4) tumor suppressor gene was downregulated in HCC cells transfected with miR183[41]. Wanget al[42]showed that miR183 was overexpressed in HCC Bel-7402/5-Fu (resistant to 5-FU) cells. P-gp and MRP2 protein expression was also increased in Bel-7402/5-Fu cells (Table 1), and the suppression of miR183 in these cells diminished P-gp and MRP2 protein levels. In addition, the authors established that high levels of miR183 induced the expression of both transporters in control cells. So, these results indicate that miR183 modulates P-gp and MRP2 expression.
Thus, a large amount of evidence points that many miRNAs are deregulated in HCC and some of them are involved in chemoresistance. Nevertheless, until the recent years the precise mechanisms underlying miRNA-induced drug resistance in this malignancy have not been fully understood. Many efforts have been targeted to decipher how aberrantly expressed miRNAs affect tumor cell proliferation or apoptosis pathways. Here, we investigated thoroughly the recent findings that reveal that certain miRNAs regulate ABC transporter expression by specifically targeting, for instance,ABCB1and/orABCC1genes. Most of these miRNAs are found downregulated in HCC tissues and cells. As a consequence, P-gp and/or MRP1 expression levels increase and intracellular therapeutic drug accumulation decrease,making HCC cells less sensitive to death (Table 1). Altogether, these findings indicate that miRNAs have a key role in ABC transporter-mediated drug resistance in HCC.
AUTOPHAGY IN THE DEVELOPMENT OF HCC CHEMORESISTANCE
Autophagy is an evolutionary conserved mechanism that involves proteolytic degradation of cytosolic components at the lysosome through lysosomal enzyme action that facilitate degradation of sequestered products[43]. It occurs mainly as a response to cellular stress (infection, hypoxia,etc.) and its leading function is to grant nutrients for cellular functions and to remove unwanted material from the cytosol such as damaged organelles, acting as a cytoprotective system[44].
Autophagy includes five phases: initiation, elongation and autophagosome formation, fusion, and autolysosome formation. This process begins with the development of an isolated membrane, called phagophore, which origin is controversial. It expands and engulfs intracellular organelles or protein aggregates(for example) in a double-membrane vesicle called autophagosome. Then, maturation occurs when autophagosome fuses with a lysosome to form an autolysosome,promoting the degradation of the inside contents. Finally, aminoacids, fatty acids, and nucleotides are transported to the cytoplasm, so they can be re-used by the cell[43].Thus, autophagy works as a recycling machinery that removes non-functional proteins and organelles.
The molecular pathway of autophagy has been thoroughly reviewed elsewhere[45,46].Briefly, the initial phase is driven by the unc-51-like autophagy activating kinase(ULK) complex and the class III phosphatidylinositol 3-kinase (PtdIns3K) complex.The last complex produces phosphatidylinositol 3-phosphate for recruitment of other factors to the phagophore, and contains the key autophagy regulators vacuolar protein sorting 34 (VPS34), VPS15, Beclin 1 and activating molecule in Beclin 1-regulated autophagy (AMBRA1). Downstream, the autophagy-related gene 8 (ATG8)and ATG12 systems (two ubiquitin-like conjugation systems) mediate vesicle expansion. The E1-like protein ATG7 is required for activation of ATG8 [light chain 3(LC3) in mammals] and ATG12. ATG8/LC3 is subjected to proteolysis and then is covalently attached to the lipid phosphatidylethanolamine (in mammalian cells, the precursor form is LC3-I and LC3-II is the lipidated one), by which it associates with the phagophore membrane. Thus, autophagy can be detected biochemically (by assessing LC3-II generation) or microscopically (by observing LC3 puncta formation,indicative of LC3 redistribution to the autophagosomes under development). The pathway includes other proteins such as ATG9, factors required for autophagosome-lysosome fusion (e.g., lysosomal-associated membrane protein 2),vacuolar permeases that mediate amino acid efflux from the lysosome, and lysosomal enzymes involved in cargo degradation[45,46].
In normal liver, damaged mitochondria and mutated cells are removed through autophagy, and this mechanism suppresses tumor initiation. But once the tumor is established, autophagy acts as a pro-oncogenic factor, as it promotes tumor growth metastasis and resistance to therapeutic drugs (revised in[47,48]). Actually, inhibition of autophagy in HCC cells incubated with sorafenib and bevacizumab enhanced cell lethality[49,50]. Moreover, autophagy modulation participated in oncolytic virotherapy in a liver cancer stem cell model[51]. Thus, appropriate autophagy regulation could effectively suppress HCC growth and metastasis. In this section, we describe the recent findings concerning the effect of miRNAs in autophagy and chemoresistance in HCC.
miR26a/b
This molecule is upregulated in some malignant cancers such as glioma and glioblastoma. On the other hand, it is downregulated in some bladder tumors, breast cancer cell lines and tissues, and anaplastic thyroid carcinoma, among others[52].Particularly in HCC, miR26a/b is downregulated and its overexpression was shown to reduce HepG2 and MHCC97-H HCC cell proliferation, migration and invasion[53].Thus, miR26a/b might play a suppressive role in liver tumor progression.
Jinet al[54]found that DOX treatment induced autophagy and reduced miR26a/b levels in HepG2 cells. Further, in DOX-resistant HepG2 cells this miRNA was also downregulated. In both cell lines, incubation with autophagy inhibitors resulted in the upregulation of miR26a/b, whereas the opposite effect was observed when an autophagy inducer was used. This implies that autophagy can modulate the expression of this miRNA[54]. Remarkably, miR26a/b overexpression in resistant cells sensitized them to apoptosis induced by DOX. Moreover, when miR-26a/b was combined with DOX treatment, miR-26a/b further improved the therapeutic effect of this drug on tumor growthin vivo. Besides, the authors performed bioinformatics analysis and reporter gene assays and found that ULK1 (a protein that participates in the initial stage of autophagy) is a target of miR26a/b in HCC cells (Table 2). The overexpression of miR26a/b induced the reduction not only of ULK1, but also of another proteins involved in autophagy (Beclin-1, ATG7 and LC3-II), on the contrary,downregulation of miR26a/b induced the increase of the formers. Thus, miR26 modulates autophagy (suppressing ULK1 expression) in order to promote apoptosis and sensitize HCC to chemotherapy. The authors propose that the combination of miR26a/b with chemotherapy could introduce a new strategy to overcome cancer[54].
miR199a-5p
In triple negative breast cancer cells this miRNA is downregulated, and suppresses cell migration and invasion[55]. On the other hand, it is upregulated in osteosarcoma cells and patient tissues and its knock-down brings about a reduction in cell proliferation and tumor growth[56]. In HCC cells and tissues, miR199a-5p is frequently downregulated and its overexpression inhibits cell proliferation, migration and invasionin vitroandin vivo[57]. Moreover, low miR199a-5p expression in HCC patients was associated with poor prognosis. Interestingly, cisplatin treatment decreased miR199a-5p levels and induced autophagy in two HCC cell lines, HuH-7 and HepG2.However, when the expression of miR199a-5p was forced, cisplatin-induced autophagy was inhibited and cisplatin-induced decrease of cell proliferation was increased. Accordingly, it was found that miR199a-5p interacts with the 3'UTR region ofATG7transcript (Table 2). Thus, cisplatin,viamiR199a-5p downregulation,increases drug resistance by inducing autophagy in HCC cells[58].
miR101
This miRNA is involved in the development and progression of oral squamous-cell cancer. In these tissues, it is downregulated and related to patient poor prognosis[59].In non-small cell lung cancer, it is almost undetectable and this absence is related to lymph node metastasis and poor prognosis of patients[60]. Further, in HCC it was observed that patients with distant metastasis had lower levels of miR101, and the downregulation of this miRNA correlated with adverse prognosis. Besides, lentivirusdelivered miR101 avoided tumor growth and metastasis in a HCC murine model[61].
In a study performed with HepG2 HCC cells, it was shown that miR101 induced apoptosis upon cisplatin treatment. By using cells transfected with a sequence of RNA that mimics miR101 it was observed that proteins involved in the phagosome formation stathmin 1 (STMN1), RAS related protein (RAB5A), autophagy-related 4D cysteine peptidase (ATG4D) and mTOR were miR101 targets (Table 2). On the other hand, the use of a sequence of RNA that specifically binds and inhibits miR101 increased the mRNA levels of the four genes. Moreover, miR101 inhibitor induced much more phagosome formation than miR101 mimics, showing that the absence of the miRNA induces autophagy. Besides, it was also demonstrated that in the presence of cisplatin, cells with lower levels of miR101 showed less apoptosis than the ones transfected with miR101 mimics. In conclusion, miR101 inhibits autophagy and increases apoptosis induced by cisplatin in HCC cells[62].
miR216b
miR216b is downregulated in colorectal cancer compared with normal tissues. High expression of this miRNA inhibited cell proliferation, migration and invasion in this type of cancer through targeting the expression of the oncogene serine-arginine protein kinase 1 (SRPK1)[63]. In cervical cancer tissues and cells it is also downregulated. Its overexpression inhibited cell proliferation in cultured HeLa cells,suggesting a possible tumor suppressor activity in this type of cancer[64]. In HCC patient plasma and tissues it was also found downregulated compared with healthy volunteers and non-tumor adjacent liver tissue, respectively. Further, low expression of miR216b correlated with patient poor prognosis[65]. In HepG2 and SMMC-7721 cells,low levels of miR216b stimulated cell proliferation, migration and invasion, while its overexpression produced the opposite effect[65]. Besides, miR216b levels in BEL-7402 HCC cells inversely correlated with the expression levels of metastasis associated lung adenocarcinoma transcript 1 (MALAT1), an oncogenic long non-coding RNA(LncRNA) that is generally upregulated in this tumor and can modulate chemosensitivity[66]. By performing gene reporter assays, it was shown that miR216b has two binding sites inMALAT1. BothMALAT1-siRNA and miR216b mimics inhibited autophagy in BEL-7402/5-FU (BEL-7402 cells 5-FU resistant) cells, and autophagy inhibition significantly increased 5-FU-induced cell apoptosis (Table 2).Moreover, these treatments decreased the IC50of 5-FU, DOX, and mitomycin in BEL-7402/5-FU cells[66]. These results revealed the relationship between autophagy,MALAT1and miR216b, contributing to HCC chemosensitivity.
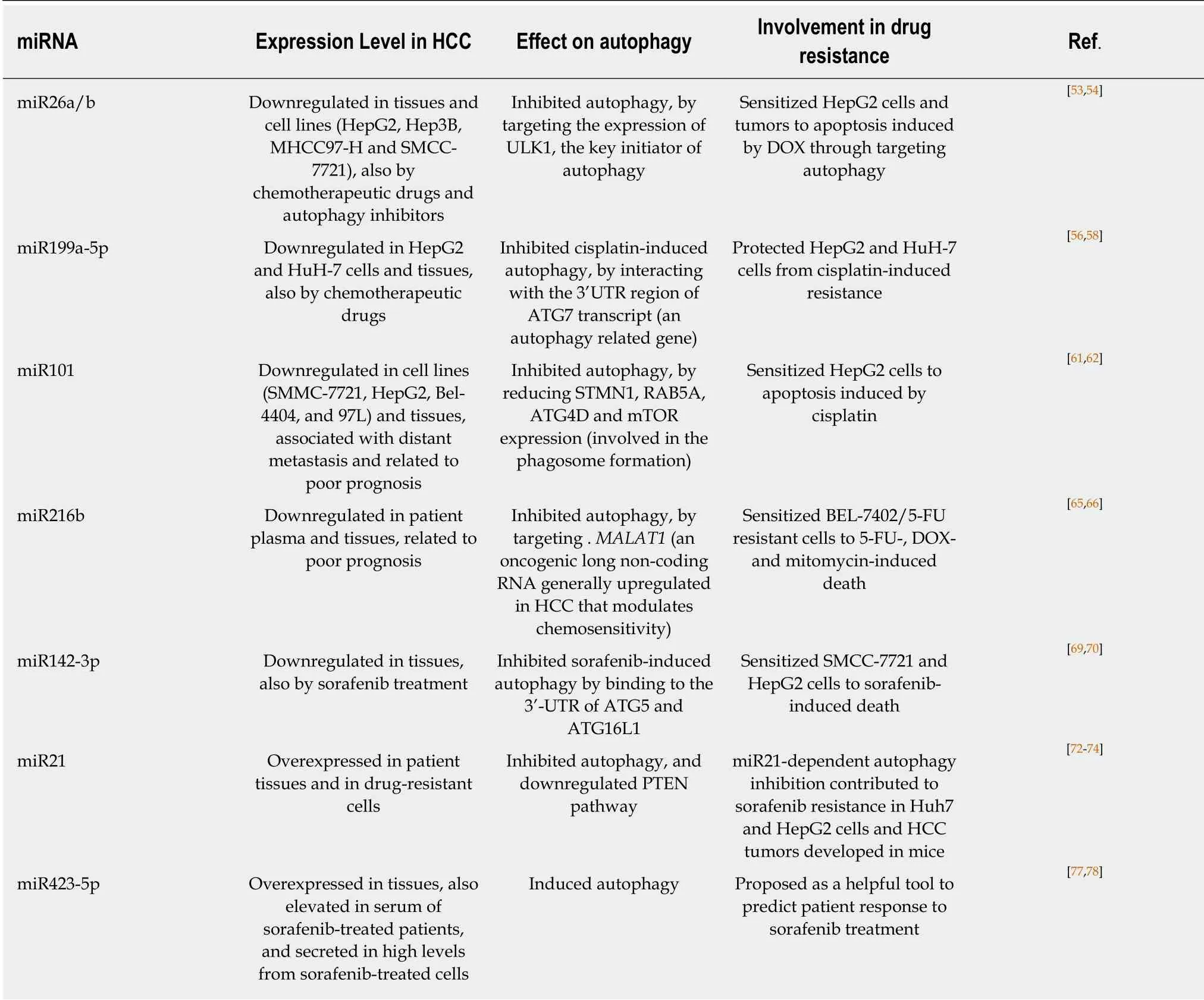
Table 2 miRNAs and autophagy in hepatocellular carcinoma cell drug resistance
miR142-3p
This miRNA is downregulated in non-small cell lung cancer (NSCLC) tissues and cell lines. At high levels, it prevents tumorigenesis inhibiting the expression of one of the high mobility group protein superfamily, HMGB1, involved in cancer cell migration and invasion[67]. Besides, this miRNA is diminished in ovarian cancer tissues and cell lines, and its downregulation is related to poor differentiation. Further, it was demonstrated that overexpression of miR142-3p inhibited proliferation and chemoresistance in ovarian cancer cells[68]. In HCC tissues it is also downregulated,and the expression levels decreased as the disease progressed. miR142-p3 overexpression inhibited BEL-7402 and SMMC-7721 HCC cell migration and invasion,thus, it was suggested to suppress metastasis[69].
Zhanget al[70]studied the role of miR142-3p in sorafenib resistance in SMMC-7721 and HepG2 HCC cells. They showed that cells incubated with this drug presented a higher autophagic flux than control cells in a dose- and time-dependent manner.When autophagy was inhibited, cells become sensible to the drug. They also showed that miR142-3p was downregulated in HCC cells under sorafenib treatment.Furthermore, it was shown that high levels of the miRNA negatively regulated sorafenib-induced autophagy in HCC cells by binding to the 3'-UTR of ATG5 and ATG16L1 mRNAs (both part of the autophagy machinery) (Table 2). Therefore, these data indicate that miR142-3p downregulation induces autophagy by increasing the level of ATG5 and ATG16L1, and thus promoting sorafenib resistance in HCC[70].
miR21
This miRNA is frequently upregulated in cancer cell lines and human tumors. It is very important in oncogenic processes as it is associated with high proliferation,reduced apoptosis, metastasis potential and invasion[71]. In HCC it is also overexpressed and it was suggested to be related to tumor progression in patients[72].Treatment with an oligonucleotide anti-miR21 led to the loss of viability, induction of apoptosis and necrosis in different HCC cell lines. Further, anti-miR21 also diminished cell migration and suppressed clonogenic growth[73].
Heet al[74]developed sorafenib-resistant HuH-7 and HepG2 HCC cells and found that miR21 was upregulated. Interestingly, drug-sensitive parental cells transfected with miRNA21 mimics also became resistant. It was also demonstrated that sorafenib upregulated the expression of two key autophagic proteins (LC3-II and Beclin-1), but the expression level of both proteins in parental cells was higher than in drugresistant ones. Besides, parental cells showed more acidic vesicular organelles than resistant cells. These results indicated that autophagy is more activated in parental cells than in the resistant ones. Interestingly, autophagy induction with rapamycin inhibited sorafenib-resistant cell growth, indicating that this process increases sensitivity to sorafenib in resistant cells[74]. Further, inhibition of miR21 by using an antisense miRNA oligonucleotide, re-sensitized resistant cells to sorafenib by promoting autophagy and stimulating apoptosis. On the other hand, miR21 mimics reduced autophagy, apoptosis, and the expression of phosphatase and tensin homologue (PTEN) protein (a known target of miR21) in parental cell lines (Table 2).Finally experiments performed with resistant cell-derived tumors established in mice showed that sorafenib administration plus intratumoral injection of anti-miR21 oligonucleotides induced a reduction in tumor size. Moreover, sorafenib administration downregulated PTEN, upregulated LC3-II and Beclin-1 expression in tumor lysates, and increased the activation of p-AKT. While anti-miR21 induced the upregulation of PTEN, LC-II and Beclin-1, and reduced expression of p-AKT[74].Therefore, acquired sorafenib resistance might be mediated by the overexpression of miR21 and the inhibition of autophagy, probably by regulating the PTEN/AKT pathway.
miR423-5p
This miRNA, overexpressed in glioblastoma, was shown to enhance angiogenesis,tumor cell growth and invasion[75]. In gastric cancer, miR423-5p was also upregulated,and this overexpression favored cell cycle progression and decreased cell migration and invasion[76]. Particularly in HCC tissues it is upregulated, and high levels of this miRNA constitute a factor that increases cell invasiveness in HCC cells[77].
Interestingly, sorafenib treatment increased miR423-5p levels in conditioned media of HepG2 and HuH-7 HCC cells and in serum of patients, suggesting a potential role of miR423-5p as a marker of sorafenib response in HCC patients. Transfection of HCC cells with an anti-miRNA diminished LC-II and ATG7 protein levels (an autophagic vacuole formation protein). Accordingly, transfection with miR423-5p mimic induced vacuole and autophagosome formation (Table 2), and the opposite was observed with the miRNA inhibitor. In conclusion, it was suggested that this miRNA has a biological role in autophagy in HCC and it was proposed as a helpful tool to predict patient response to sorafenib treatment[78].
Thus, most miRNAs described here target the expression of autophagy-related genes, inhibiting autophagy and acting as tumor suppressors. However, in HCC tissues and cells many of these miRNAs are downregulated, or their levels are reduced after therapeutic drug treatment. As a consequence of this downregulation,miRNAs do not inhibit autophagy and tumor growth and resistance is favored (Table 2).
CONCLUSION
miRNAs are deregulated in HCC cells and tissues, and they play crucial roles in the development of chemoresistance. Emerging studies point that the modulation of ABC transporter and/or autophagy-related gene expression could be determinant for HCC cell survival under chemotherapeutic drug treatment. Our knowledge about the precise mechanisms regarding miRNA involvement in resistance will lead us to find new ways of making HCC treatment more effective. The development of miRNAs,miRNAs mimics or anti-miRNA with long half lives and their use in combination with chemotherapeutic drugs could be a powerful option. Even so, there are still difficulties to overcome prior the possibility of being able to use miRNAs in clinical trials, for instance, the right delivery system. Nowadays, much research is being conducted on the use of nanoparticles to get over this trouble. Undoubtedly, more insights on the biological processes, signaling pathways and/or molecular mechanisms regulated by miRNAs are needed. Anyway, miRNA-based therapy together with conventional chemotherapeutic drugs has a great future in cancer therapy.
 World Journal of Hepatology2019年4期
World Journal of Hepatology2019年4期
- World Journal of Hepatology的其它文章
- Endoscopic ultrasound guided liver biopsy for parenchymal liver disease
- Beneficial effects of losartan or telmisartan on the local hepatic renin-angiotensin system to counter obesity in an experimental model
- Being accompanied to liver discharge clinic: An easy measure to identify potential liver transplant candidates among those previously considered ineligible
- Effectiveness of venous thromboembolism prophylaxis in patients with liver disease
- Nonalcoholic fatty liver disease prevalence in an Italian cohort of patients with hidradenitis suppurativa: A multi-center retrospective analysis
- Leukocytoclastic vasculitis caused by hepatitis C virus in a liver transplant recipient: A case report