
含有喹啉结构片段的杂合体及其抗耐药疟疾活性(一)
2019-06-16 06:09文胜编写徐志审校
国外医药(抗生素分册) 2019年6期
文胜 编写,徐志,2 审校
(1 武汉弗顿控股有限公司,武汉 430073;2 湖北德信辰科技有限公司,武汉 430080)
1 前言
疟疾主要是由恶性疟原虫引起的一类高传染性、致死性疾病。全球约一半人口生活在感染疟疾的阴影之下,每年新增超2亿感染病例,数十万人因此丧命。据世界卫生组织(WHO)估计,2017年全球疟疾新发病例仅比2010年少2000万,且2015—2017年间在疟疾的防控方面并未取得实质性进展,疟疾的防控形势依然严峻。

图1 四类喹啉杂合体
抗疟疾药物对疟疾的防控至关重要,其中喹啉类药物如奎宁(QN)、二茂铁喹(FQ)、氯喹(CQ)、阿莫地喹(AQ)、甲氟喹(MQ)和伯氨喹(PQ)是治疗疟疾的基石药物。这类药物可通过抑制疟色素形成或插到DNA双螺旋之间形成稳定的复合物阻止DNA的复制和RNA的转录,进而杀灭疟原虫。然而,随着这类药物的长期广泛使用,疟原虫对其产生了严重的耐药性。其中,耐氯喹(CQR)恶性疟疾在院内最为常见。目前,WHO推荐基于青蒿素的联合疗法(ACTs)作为治疗疟疾包括耐喹啉疟疾的标准疗法,但该疗法存在生物利用度低、复发率高、溶解性差、价格昂贵等缺点。不仅如此,耐青蒿素疟疾已然出现。因此,亟需研发新型抗疟疾药物。
杂合体具有克服耐药性、增强抗疟疾活性和降低毒副作用的潜力,且目前多个杂合体药物处于临床研究阶段,有望于不久的将来为人类健康服务。喹啉类化合物具有优秀抗疟疾活性,显然,将喹啉母核与其它抗疟疾活性药效团杂合是获得新型抗疟疾药物的有效途径之一。
自2010年以来,科学家报道的喹啉类杂合体大体可分为以下四类:① I型杂合体,即喹啉-青蒿素(及其衍生物)杂合体;② II型杂合体,即喹啉-合成过氧化物杂合体;③ III型杂合体,即喹啉-耐药逆转剂杂合体和IV型杂合体,即喹啉-新型抗疟疾药效团杂合体(图1)。本文将重点介绍I型~III型喹啉类杂合体的体内外抗耐药疟疾活性,并探讨构-效关系(SAR),以期为进一步研究提供理论支持。
2 喹啉-青蒿素(及其衍生物)杂合体(I型杂合体)
青蒿素(ART)及其半合成衍生物如双氢青蒿素(DHA)、蒿甲醚、蒿乙醚和青蒿琥酯等不仅可作用于所有的可感染人类疟原虫,对红内期和有性生殖期恶性疟原虫高度有效,而且还具有起效快和毒副作用低等诸多优点。目前,ACTs是世界范围内治疗疟疾最有效的手段,对药敏型和耐药型疟疾均有效,极大地降低了疟疾的发病率和死亡率。然而,ART及其半合成衍生物具有生物利用度低、药代动力学性质差、复发率高和价格昂贵等缺点,且耐ACTs疟原虫依然出现。为克服上述缺陷,迫切需要对此类药物进行修饰。喹啉(作用于肝期)和ART(作用于血期)的作用机制大相径庭,故将喹啉和ART及其衍生物糅合到一个分子中可能会获得新型抗疟疾候选物。
喹啉-双ART杂合体1(图2)具有良好的体外抗氯喹敏感型(CQS)D10和CQR Dd2恶性疟原虫活性,半数抑制浓度(IC50)为5.31~205.42nmol/L。细胞毒性研究结果表明,这类杂合体对CHO细胞系的毒性较低(IC50:0.68~74.82μmol/L)。耐药性指数(RI:IC50(Dd2)/IC50(D10))和选择性指数(SI:IC50(CHO)/IC50(D10))分别为2~6和61~3813。SAR显示喹啉和ART之间的连接子与活性息息相关:对烷基连接子而言,丙基(n=2)>乙基(n=1)>>丁基(n=3),而用哌嗪基乙基取代烷基并不能改善活性。代表物1b(IC50:5.31和28.43nmol/L)对所测CQS D10和CQR Dd2恶性疟原虫的活性是CQ(IC50:21.54和157.90nmol/L)的4.0和5.5倍,抗CQS D10活性与DHA(IC50:5.11nmol/L)相当。在鼠疟感染的小鼠模型中,给药杂合体1a(静脉注射给药/ip 2.5mg/kg或口服给药/po 50mg/kg)或1d(ip 7.5mg/kg或po 25mg/kg)在d5小鼠的疟原虫血症<1%,而青蒿琥酯(30mg/kg ip)对照组原虫血症为3%。杂合体1a和1d(1.4和2.1mg/kg)的ip平均半数有效剂量(ED50)与青蒿琥酯(<1mg/kg)相当,但po ED50(20和13mg/kg)则显著高于青蒿琥酯(1.8mg/kg)。快速药代动力学分析结果表明,这类杂合体的抗恶性疟原虫活性可能归因于其活性代谢产物,而非杂合体本身,这一点得到了分子对接实验的进一步佐证。
7-氯喹林-ART杂合体2具有良好的抗CQS D10和CQR Dd2恶性疟原虫活性,IC50分别为12.18~201.38nmol/L和17.12~275.99nmol/L。尽管绝大多数杂合体的活性高于CQ(IC50:21.54和157.90nmol/L),但均弱于DHA(IC50:5.11和2.09nmol/L)。这类杂合体的RI(1~2)在1左右,提示可能具有全新的作用机制。SAR显示,连接子的长度与这类化合物的抗CQR Dd2活性相关,且两个碳二胺对活性有利。与相应的游离碱相比,草酸盐的活性更高。代表物2d(IC50:12.18和17.12nmol/L)的抗CQR Dd2恶性疟原虫活性是CQ的9.2倍,且该化合物(IC50:3.39μmol/L)对CHO的毒性较低,SI高达279。在鼠疟感染的小鼠模型中,杂合体2a和2d在ip给药剂量为15mg/kg和po给药剂量为50mg/kg时可完全治愈感染小鼠,而青蒿琥酯仅可在ip给药剂量为30mg/kg和po给药剂量为80mg/kg时获得类似的结果。杂合体2a(po给药剂量为20mg/kg时的达峰时间为23min,最大浓度为141ng/mL)的某些药代动力学性质与DHA(po给药剂量为10mg/kg时的达峰时间为48min,最大浓度为142.2ng/mL)相当,且消除速率是DHA(6.1L/min/kg vs 1.19L/min/kg)的5倍,但DHA(ip)的半衰期(t1/2:25min vs 3.9min)、药时曲线下面积(AUC, 8748 ng·min/mL vs 3463ng·min/mL)和分布容积(353.5L/kg vs 34L/kg)均优于该杂合体。
7-氯喹啉-ART杂合体3(IC50:19~35nmol/L)的抗CQS D10和CQR K1恶性疟原虫活性与ART(IC50:23和14nmol/L)相当,抗CQR K1活性是CQ(IC50:219nmol/L)的≥9.5倍。杂合体3a~c的活性相当,提示连接子的长度和R位的取代基对活性影响不大。用酯基取代醚所得的杂合体4(IC50:18.7~55.6nmol/L)对CQS D10和CQR K1恶性疟原虫也具有良好的活性,且延长连接子的长度对抗CQR Dd2活性不利。杂合体3(0.88, 0.60和0.70)的RI<1,提示这类杂合体与CQ无交叉耐药性,具有治疗耐药疟疾的潜力。进一步研究显示,这类杂合体可能同时具有CQ和ART的作用机制,故发生耐药性的几率较低。
PQ-ART杂合体5(IC50:5.1-523nmol/L)对红内期CQR W2恶性疟原虫和红外期伯氏疟原虫均显示出良好的活性,是PQ(IC50:3300和7500nmol/L)的14~647倍,而抗红内期CQR W2恶性疟原虫活性与ART (IC50:8.2nmol/L)相当。含有苯基的杂合体5a,b(IC50:9.1和5.1nmol/L)的抗红内期CQR W2恶性疟原虫活性高于化合物5c(IC50:12.5nmol/L),提示苯基的引入对活性有利;苯酰胺杂合体5b(IC50:5.1和67nmol/L)的活性优于相应的胺基衍生物5a(IC50:12.5和155nmol/L),提示酰胺比胺基对活性有利。在感染鼠疟的小鼠模型中,杂合体5a的体内活性如治愈率和存活率优于单独给药ART或PQ与ART复方制剂。显然,这类可同时作用于红内期和红外期杂合体在治疗疟疾尤其是耐药疟疾领域有着广泛的应用前景。
含有ART、CQ和PQ结构片段的ART-吖啶杂合体6对CQS NF54和CQR Dd2恶性疟原虫的IC50为2.6~429.9nmol/L,且除化合物6c之外的所有杂合体活性均优于CQ(IC50:18.5和271.7nmol/L),但弱于DHA(IC50:<7和7nmol/L)。SAR显示,连接子与活性息息相关,且烷基二胺优于哌嗪基。杂合体6a(IC50:2.6和35.3nmol/L)对所测CQS NF54和CQR Dd2恶性疟原虫的活性最高,是CQ的7.1和12.1倍。然而,该杂合体的RI(13.6)与CQ(14.7)相当,提示该杂合体可能与CQ具有相似的作用机制。
青蒿琥酯-吲哚并喹啉杂合体7~9抗CQS NF54和CQR K1恶性疟原虫的IC50为0.41~3.21nmol/L,高于对照药CQ(IC50:9.4和209.5nmol/L)和ART(IC50:4.3和2.8nmol/L)。细胞毒性实验结果表明,这类杂合体(IC50:695.0~3,7183nmol/L)对L6细胞的毒性较低,选择性良好。值得一提的是,此类杂合体的RI低至0.27~0.93,提示这类化合物与CQ无交叉耐药性。与杂合体7相比,其无取代衍生物8和异构体9显示出相似的活性。对杂合体7而言,向R1、R2和R3位引入供电子基或吸电子基均活性不利。代表物7a(IC50:0.45和0.42nmol/L)对所测CQS NF54和CQR K1恶性疟原虫的活性是CQ和ART的9.5~498.8倍,SI>3641,优秀的体外活性和良好的安全性使得该杂合体极具进一步开发前景。
3 喹啉-合成过氧化物杂合体(II型杂合体)
ART类化合物抗疟疾作用主要是通过二价铁裂解青蒿素的过氧桥结构产生大量的自由基,而自由基与疟原蛋白结合,作用于疟原虫的膜系结构,进而诱导疟原虫死亡。1,2,4-三氧环己烷和1,2,4,5-四氧环己烷均含有过氧结构片段,是ART结构骨架的有效替代结构单元。某些含有1,2,4-三氧环己烷或1,2,4,5-四氧环己烷药效团的化合物如OZ227、OZ439和CDRI-97/78等对耐药疟疾具有潜在的疗效,目前已用于临床或处于临床评价阶段。显然,将喹啉与过氧化物药效团杂合是获得对耐药疟疾具有优秀活性的有效途径。
四氧环己烷-PQ杂合体10(图3)对血期CQR W2恶性疟原虫具有良好的活性,IC50为21.1~45.2nmol/L,活性是PQ(IC50:3300nmol/L)的73~156倍。其中,化合物10a(IC50:538nmol/L)、10c(IC50:604nmol/L)和10d(IC50:330nmol/L)对伯氏疟原虫也显示出优秀活性,是PQ(IC50:3300nmol/L)的12.4~22.7倍。连接子与活性息息相关,且贡献顺序为-NH- > -CONH- > -CH2CONH- > -CH2NH-,提示短链连接子对活性有利。在绿色荧光蛋白(GFP)表达的伯氏疟原虫ANKA感染的C57Bl/6J雄性小鼠模型中,所有杂合体在浓度为25μmol/(L·kg)均可显著地降低受感染蚊子(86.7%~100%)和卵囊(98.2%~100%)数量,提示这类杂合体可有效地阻断疟原虫向蚊子的传播。代表物10a可完全抑制幼蚊卵囊内疟原虫,活性优于PQ (99%)。显然,这类杂合体在疟疾的防治领域可能大有作为。
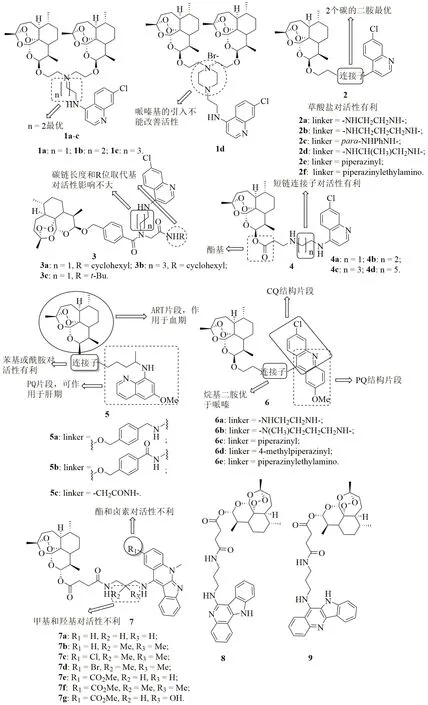
图2 杂合体喹啉-ART(及其衍生物)杂合体1~9的化学结构
为进一步研究连接子对活性的影响,药物化学家进一步评价了不同连接子连接的四氧环己烷-PQ杂合体11~13的抗红内期CQR W2恶性疟原虫和肝期伯氏疟原虫的体外活性。初步研究结果表明,除化合物13c,d外的所有杂合体对红内期CQR W2恶性疟原虫均显示出极高的活性,EC50在纳摩尔水平。SAR研究结果表明,四氧环己烷与PQ之间的连接子和喹啉C-5位取代基对杀血期疟原虫裂殖体活性至关重要。杂合体11(EC50:15~204nmol/L)和12(EC50:19~176nmol/L)的活性高于13(EC50:287~5670nmol/L),提示四氧环己烷与PQ之间的烷基连接子对高活性至关重要。对杂合体11而言,含有直链烷烃连接子的化合物11j~l(EC50:15~17nmol/L)活性优于相应的支链杂合体(如11c, EC50:47nmol/L)。杂合体12的抗恶性疟原虫活性与化合物11处于同一水平,提示胺基与酰胺对活性的贡献相当。此外,向喹啉苯环的C-5位引入吸电子基比引入供电子基对活性更有利。杂合体11(EC50:1.11~4.44μmol/L)的抗肝期伯氏疟原虫活性高于杂合体12(对宿主有毒性),且前者对哺乳动物CaCo-2细胞系未显示出任何毒性,CC50>50μmol/L。代表物11k(EC50:15nmol/L和1.11μmol/L)抗CQR W2恶性疟原虫活性与ART(EC50:10nmol/L)相当,而抗伯氏疟原虫活性则是CQ(EC50:7.5μmol/L)的6.7倍。由此可见,将喹啉与过氧化物药效团杂合的策略可获得具有多重作用机制的抗疟疾候选物。
7-氯喹林-四氧环己烷杂合体14(IC50:3.906~4.814μmol/L)的抗CQR RKL-9恶性疟原虫活性与杂合体15(IC50:3.906~5.629μmol/L)相当,但均弱于对照药CQ(IC50:0.393μmol/L)。SAR研究结果显示,7-氯喹啉与四氧环己烷之间的连接子及四氧环己烷上的取代基如二甲基或环烷基对抗CQR RKL-9恶性疟原虫活性影响较小,故二者非今后优化的重点。
7-氯喹啉-二茂钌-三氧环己烷杂合体16(图4, IC50:65.33和62.00nmol/L)对CQR K1和Dd2恶性疟原虫具有良好的活性,与ART(IC50:17.43和18.67nmol/L)相当,而是CQ(IC50:1,086和521.67nmol/L)的16.6和8.4倍。此外,杂合体16(IC50:1.48和1.55μmol/L)对L6和MRC5哺乳细胞系的毒性较低,值得进一步研究。
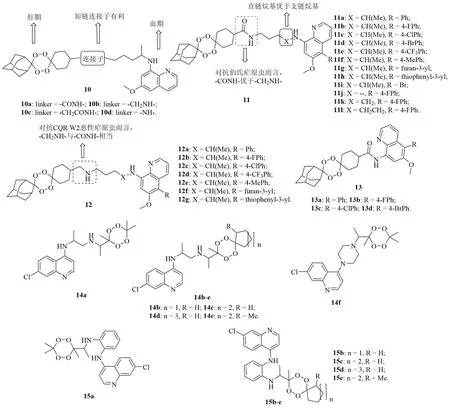
图3 喹啉-四氧环己烷杂合体10~15的化学结构
含有二茂铁结构片段的7-氯喹啉-三氧环己烷杂合体17(IC50:16~71nmol/L)对CQR FcB1和FcM29恶性疟原虫的活性与ART(IC50:10和18nmol/L)处于同一水平,而是CQ(IC50:145和735nmol/L)的3.3~45.9倍。其中,抗CQR FcB1和FcM29恶性疟原虫活性最高的杂合体17a(IC50:20和17nmol/L)在文氏疟原虫感染小鼠模型中也显示出良好的活性,即使给药剂量低至每日10mg/kg,连续给药4d时,小鼠的疟原虫血症降至检测线以下。当给药剂量为每天25mg/kg时,小鼠的平均存活时间为18d,这可能是由于在给药17~21d之间疟疾复发所致。
喹啉-过氧化物杂合体1 8(抗CQSD10和CQR W2恶性疟原虫的IC50分别为48~90nmol/L和110~340nmol/L)和19(IC50:210~940nmol/L)具有良好的抗恶性疟原虫活性,但活性弱于DHA(IC50:5和2.4nmol/L)。此类杂合体(IC50:110~500nmol/L)的抗CQR W2恶性疟原虫活性优于CQ(IC50:700nmol/L),且杂合体18的抗CQS D10恶性疟原虫活性与CQ(IC50:50nmol/L)相当。SAR研究结果表明,杂合体18的抗CQS D10和CQR W2恶性疟原虫均优于化合物19,提示喹啉环对活性至关重要。对杂合体18而言,连接子的长度对活性影响不大。除化合物18g外的所有杂合体的RI为0.5~2.6,远低于CQ(14),提示这类杂合体具有治疗耐药疟疾的潜力。不仅如此,所有杂合体(IC50:2.24~>38μmol/L)对HMEC-1细胞的毒性较低,SI为19~>760,说明这类杂合体的安全性良好。
7 - 氯喹啉- 过氧化物杂合体20(IC50:0.185~6.75μmol/L)具有中等强度的体外抗CQS 3D7和CQR W2恶性疟原虫活性,且活性远弱于对照药ART (IC50:19和19nmol/L)。杂合体20b(IC50:2.45和1.40μmol/L)的活性优于相应的双键衍生物20a(IC50:6.75和6.05μmol/L),提示将双键还原可在一定程度上提高活性。用甲氧基代替羟基可提高活性,如杂合体20c(IC50:240和185nmol/L)的抗恶性疟原虫活性优于20a,b。值得一提的是,所有杂合体的RI均<1,提示这类杂合体具有全新的作用机制。其中,代表物20c的抗CQR W2恶性疟原虫活性是CQ(IC50:420nmol/L)的2.2倍,值得进一步研究。
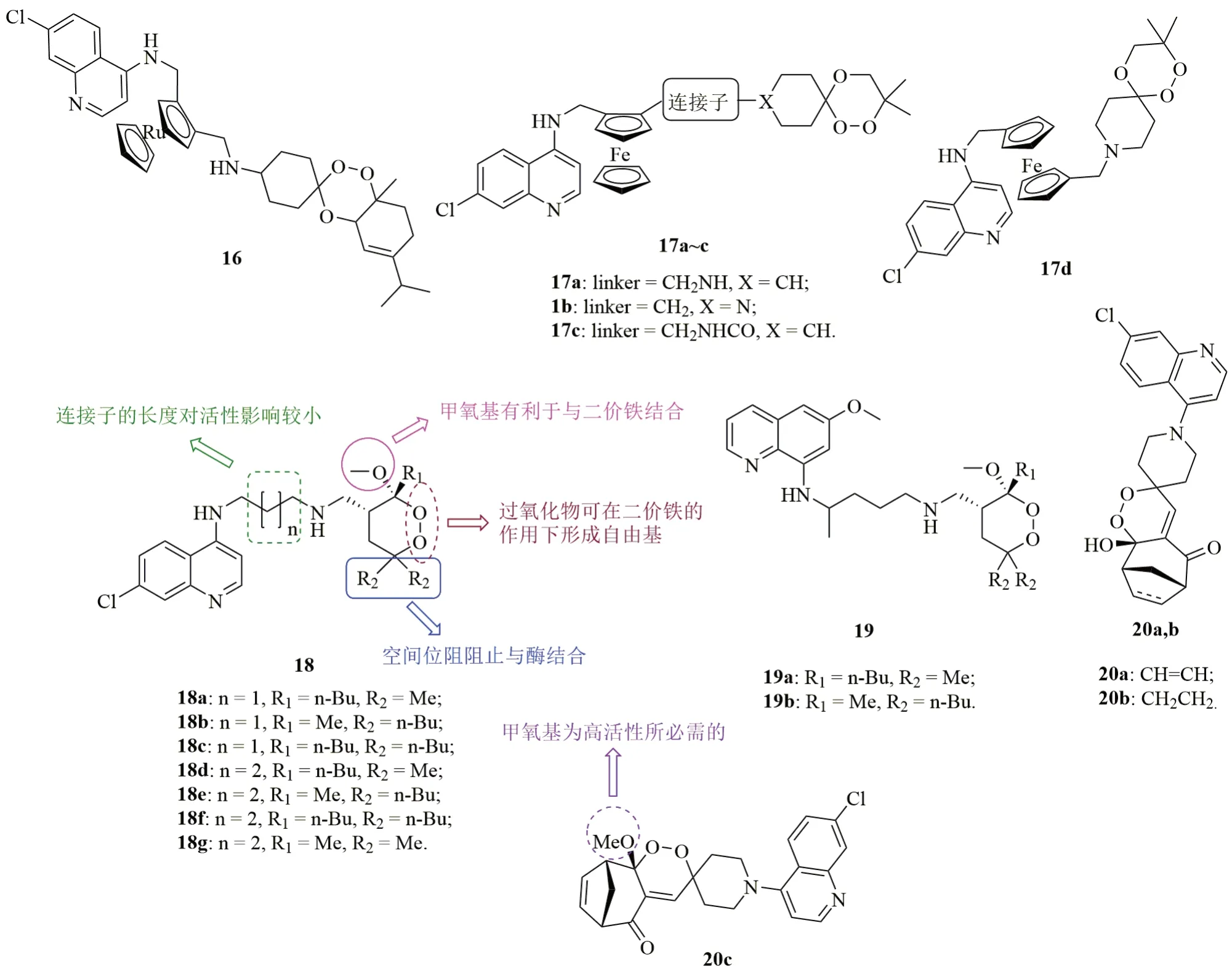
图4 喹啉-三氧环己烷/过氧化物杂合体16~20的化学结构
4 喹啉-耐药逆转剂杂合体(III型杂合体)
恶性疟原虫对CQ敏感度降低主要是由于恶性疟原虫氯喹抗性转运蛋白编码基因(PfCQRT)突变导致CQ被排出消化液泡,进而导致作用位点CQ浓度不足所致。通过将耐药逆转剂(如丙咪嗪、维拉帕米和地昔帕明等)与CQ杂合的策略可有效地干扰PfCQRT,近年来引起了药物化学家的广泛关注。
Burgess等评价了7-氯喹啉-丙咪嗪杂合体21a,b、7-氯喹啉-吡啶杂合体21c,d和7-氯喹啉-金刚烷杂合体21e(图5)的体内外抗CQS和CQR疟疾活性及细胞毒性。结果表明,所有杂合体不仅对小鼠脾淋巴细胞(IC50:0.7~130μmol/L)的毒性较低,而且对CQS D6、CQR Dd2和7G8恶性疟原虫(IC50:0.8~21nmol/L)的活性极高,SI高达132~69400。总体而言,耐药逆转剂的类型直接影响抗恶性疟原虫活性,且吡啶≥丙咪嗪>金刚烷。7-氯喹啉与耐药逆转剂之间的连接子长度也与活性相关,且长链连接子优于短链连接子。作用机制研究结果表明,这类杂合体与CQ类似可抑制疟原虫消化液泡内血红素的形成。在鼠疟感染的小鼠模型中,口服给药(4×30mg/kg)和皮下注射给药(1×30mg/kg)杂合体21d小鼠的疟原虫血症分别为<1%和0.7%,体内活性可与CQ(0.1%和0.5%)相媲美。
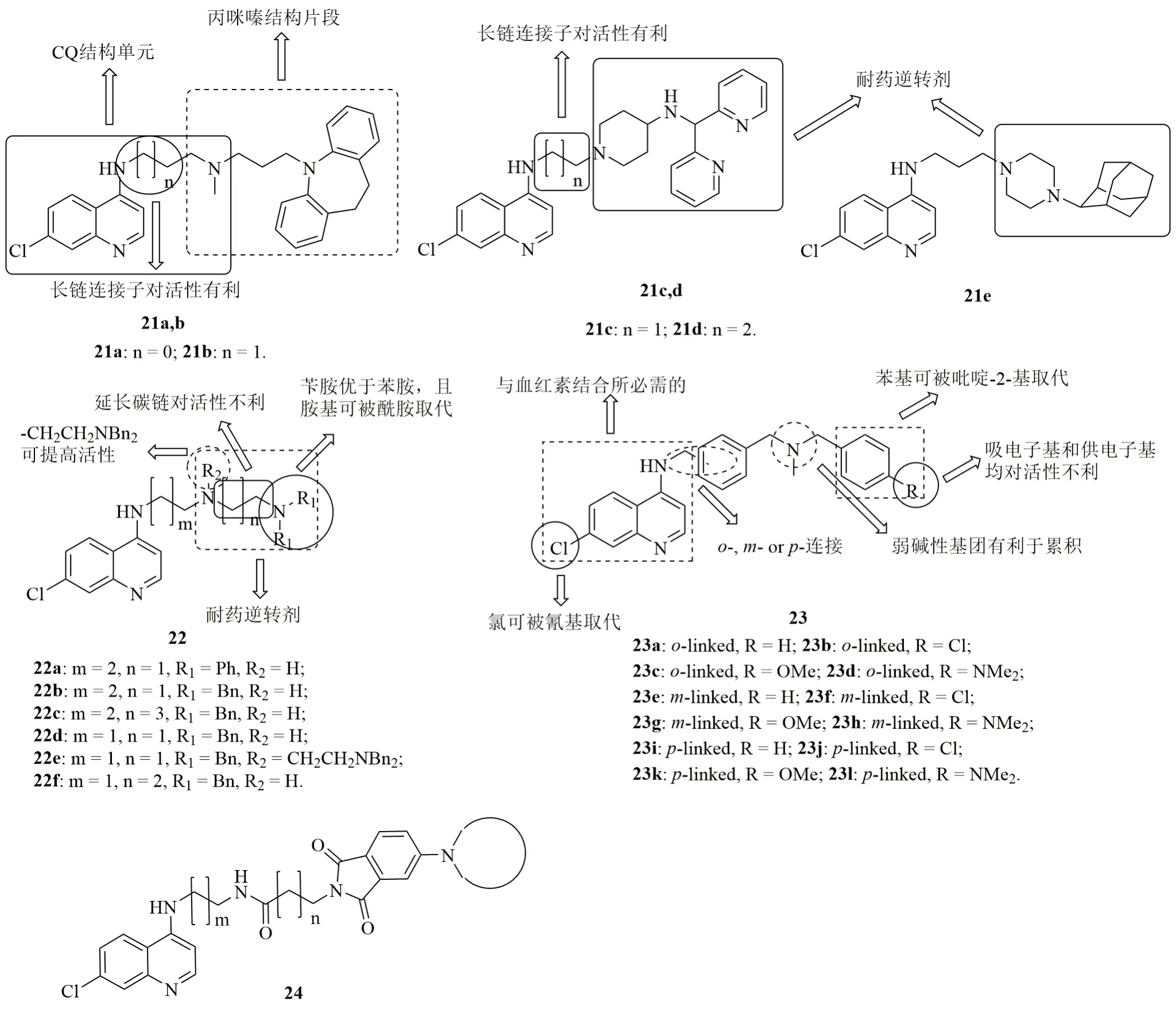
图5 喹啉-耐药逆转剂杂合体21~24的化学结构
对喹啉-耐药逆转剂杂合体22(IC50:2~34nmol/L)的体外抗CQS D6和CQR Dd2恶性疟原虫活性评价结果表明,氮原子上的取代基(R1和R2位)及连接子的长度对活性有显著影响。对R1位而言,苄基优于苯基,且取代胺基可由酰胺替换。与无取代的衍生物相比,向R2位引入-CH2CH2NBn2可提高活性。延长喹啉与耐药逆转剂之间连接子的碳链长度对活性不利,且烷基二胺连接子远优于哌嗪。代表物22e (IC50:2和5nmol/L)的抗CQS D6和CQR Dd2恶性疟原虫活性优于CQ(IC50:7和102nmol/L),且对小鼠脾淋巴细胞的毒性较低,IC50为6.2μmol/L。因此,该杂合体可作为先导物进一步优化。
邻位、间位和对位连接的7-氯喹啉-双苄甲胺杂合体23(IC50:22~1128nmol/L)的抗CQS D10和CQR K1恶性疟原虫活性与CQ相当或更优,且RI为0.6~1.3,提示这类杂合体几乎与CQ无交叉耐药性。连接方式和抗恶性疟原虫活性息息相关,且对位>间位>邻位。与无取代衍生物相比,向R位无论引入吸电子基如氯还是供电子基如甲氧基或二甲胺基均会导致活性大幅降低。进一步研究结果表明,喹啉C-7位的氯原子可被氰基取代,且双苄甲胺末端的苯基可被吡啶-2-基取代。代表物23i(IC50:22~38nmol/L)显示出优异的抗CQS D10和CQR K1、Dd2、W2和RSA11恶性疟原虫活性,抗CQS D10活性与CQ(IC50:23nmol/L)相当,而抗CQR恶性疟原虫活性则是CQ(IC50:125~170nmol/L)的>4.4倍。作用机制研究结果表明,杂合体23a、23e和23i(IC50:36~58μmol/L)具有同时抑制血红素形成和PfCQRT活性。在感染鼠疟的小鼠模型中,杂合体23a和23e(4×100mg/kg,口服给药)可在给药期内降低>99%的疟原虫血症,且未见小鼠死亡。显然,二者极具进一步研发前景。
除此之外,其它一些喹啉-耐药逆转剂杂合体如喹啉-二氧代异吲哚啉杂合体24也具有潜在的抗CQR恶性疟原虫活性,但绝大多数杂合体活性弱于相应的对照药。尽管如此,此类研究丰富了SAR,为进一步研究指明了方向。
5 结束语
疟疾是危害最严重的传染性疾病之一,与艾滋病、结核病被认为是全球最重要的三大公共卫生问题。抗疟疾药物对疟疾的防控至关重要,但随着抗疟疾药物的长期、广泛使用,疟原虫对几乎所有现有药物产生了不同程度的耐药性。因此,亟需研发新型尤其是对耐药疟疾有良好疗效的抗疟疾药物。
杂合体具有克服耐药性、增强抗疟疾活性和降低毒副作用的潜力,故将具有优秀抗疟疾活性的喹啉母核与其它抗疟疾活性药效团杂合是获得新型抗疟疾药物的有效途径之一。近年来,科学家设计合成并评价了多个喹啉杂合体的体内外抗疟疾尤其是耐药疟疾活性,并发现了若干苗头化合物。本文将着重介绍自2010年以来,喹啉-青蒿素(及其衍生物)杂合体、喹啉-合成过氧化物杂合体及喹啉-耐药逆转剂杂合体在抗耐药疟疾领域的最新研究进展,为进一步研究提供理论支持。
猜你喜欢
科学导报(2022年28期)2022-05-24
化工管理(2021年7期)2021-05-13
发明与创新(2020年5期)2020-12-20
科学导报(2020年69期)2020-11-09
环境保护与循环经济(2020年4期)2020-06-08
中国特种设备安全(2019年1期)2019-03-13
郑州大学学报(医学版)(2018年5期)2018-10-10
中国感染与化疗杂志(2018年6期)2018-01-19
中成药(2017年7期)2017-11-22
国外医药(抗生素分册)(2016年1期)2016-07-10
