
含有喹啉结构片段的杂合体及其抗耐药疟疾活性(二)
2019-06-16 06:09卢勇编写徐志审校
国外医药(抗生素分册) 2019年6期
卢勇 编写,徐志,2 审校
(1 武汉弗顿控股有限公司,武汉 430073;2 湖北德信辰科技有限公司,武汉 430080)
1 前言
近年来的研究表明,其它一些药效团如氨基酸、唑、查尔酮、肉桂酸、二茂铁、吲哚/吡咯、内酰胺、大环内酯、嘧啶、吡啶/吡啶酮和1,3,5-三嗪等药效团也具有潜在的抗疟疾活性。显然,将这些新型药效团与喹啉杂合也是获得对药敏型和耐药性疟疾均有良好疗效的抗疟新药候选物的潜在途径。
本文将综述自2010年以来喹啉二聚体、喹啉-氨基酸、喹啉-唑、喹啉-查耳酮、喹啉-肉桂酸、喹啉-二茂铁、喹啉-吲哚/吡咯、喹啉-内酰胺、喹啉-大环内酯、喹啉-嘧啶、喹啉-吡啶/吡啶酮和喹啉-1,3,5-三嗪等喹啉类杂合体即IV型杂合体在抗耐药疟疾领域的最新研究进展,并探讨了此类杂合体的构-效关系(SAR),以期为进一步研究提供理论支持。
2 喹啉-新型抗疟疾药效团杂合体(IV型杂合体)
2.1 喹啉二聚体
与相应的单核化合物相比,二聚体往往具有一些特殊的性质。特别值得一提的是,喹啉二聚体PQ目前已被临床用于疟疾包括耐药疟疾的防治。由此可见,喹啉二聚体值得进一步研究。
奎宁(QN)二聚体25a,b(IC50:514.1和297.6nmol/L)的抗氯喹敏感型(CQS) HB3恶性疟原虫活性弱于QN(IC50:108.2nmol/L),但抗耐氯喹型(CQR) P31、FCB和Dd2恶性疟原虫活性(IC50:32.0~232.1nmol/L)则优于QN(IC50:187.7~535.3nmol/L),见图6。这类二聚体的耐药指数(RI)低至0.11~0.78,提示其与氯喹(CQ)无交叉耐药性。在鼠疟感染的小鼠模型中,二聚体25b(ED50:38.2mg/kg)的体内活性优于QN(ED50:48.7mg/kg),提示这类二聚体值得深入研究。
7-氯喹啉二聚体26(IC50:24~2080nmol/L)和27 (IC50:355~2090nmol/L)具有优秀的抗CQS 3D7和CQR K1恶性疟原虫活性,但抗CQS 3D7活性弱于CQ(IC50:5nmol/L)。与二聚体27相比,化合物26的活性更高,提示哌嗪的引入对活性不利。代表物26d (IC50:24和26nmol/L)具有优异的抗CQS 3D7和CQR K1恶性疟原虫活性,且抗CQR K1活性是CQ(IC50:255nmol/L)的9.8倍。7-氯喹啉二聚体28a,b(IC50:10~190nmol/L)具有优秀的抗CQS(D10和NF54)和CQR(W2和K1)恶性疟原虫活性,且对CQR恶性疟原虫的活性(IC50:10~98nmol/L)高于抗CQS恶性疟原虫活性(IC50:83~190nmol/L),提示这类化合物可能具有全新的作用机制。化合物28a(IC50:58和10nmol/L)抗CQR恶性疟原虫活性是CQ(IC50:280和154nmol/L)的4.8和15.4倍,可作为先导物进一步研究。甾体连接的7-氯喹啉二聚体29a,b(IC50:98.19~194.74nmol/L)对CQS D6、CQR W2和耐多药TM91C235恶性疟原虫显示出潜在的活性,且对耐多药TM91C235恶性疟原虫的活性与CQ(IC50:138.82nmol/L)相当,而抗CQR W2恶性疟原虫活性则是CQ(IC50:456.20nmol/L)的4倍。
对4-胺基喹啉-2-胺基喹啉杂合体30而言,杂合体30b(IC50:0.92和37nmol/L)的抗CQS FCR-3和CQR K1恶性疟原虫活性高于化合物30a(IC50:29和370nmol/L), 提示延长连接子的长度对活性有利。杂合体30b的抗CQS FCR-3和CQR K1恶性疟原虫活性是CQ(IC50:46和570nmol/L)的50和15.4倍,且对人胚胎肺成纤维细胞MRC-5的毒性(CC50:2200nmol/L)较低,值得进一步研究。
大多数异核喹啉二聚体31显示出良好的抗CQR K1恶性疟原虫活性,IC50在纳摩尔级,且半数二聚体(IC50:350~470nmol/L)优于CQ(IC50:930nmol/L)。与无取代衍生物相比,向R位引入氯原子会导致活性的降低。对无取代衍生物而言,连接子的长度与活性正相关,即连接子越长,二聚体的活性越高。
喹啉-吖啶杂合体32(MIC:0.25~1μg/mL)的抗CQS NF54恶性疟原虫SAR研究结果显示,含有对苯二基和间苯二基连接子的杂合体活性优于相应的烷基连接子衍生物,但均弱于CQ(MIC:0.125μg/mL)。在感染CQR N-67约氏疟原虫的小鼠模型中,杂合体32b (每日4·50mg/kg, 腹腔注射给药)在给药d4出现治疗反应,但在d28无小鼠存活。
研究表明,喹啉二聚体33对所测的耐药恶性疟原虫N093、A156、A160和K239临床分离株具有良好的活性,且绝大多数二聚体的IC50在纳摩尔级。代表物33e对所测临床分离株具有广谱活性,IC50为769.6~1122nmol/L,但弱于对照药青蒿琥脂(IC50:2.6~12.68nmol/L)和CQ (IC50:22.22~121.20nmol/L)。
除此之外,某些非对称的喹啉二聚体也显示出良好的抗耐药恶性疟原虫活性,如二聚体34的抗CQR W2和耐MQ 3D7恶性疟原虫的IC50分别为71和70nmol/L,活性可与MQ和CQ(IC50:32~200nmol/L)相媲美。
2.2 喹啉-氨基酸杂合体
氨基酸可作为运输药物的载体,故将氨基酸与喹啉杂合可能能使喹啉药物更高效的达到作用靶点,进而增强对耐药疟疾的活性。Kuar等评价了三个系列PQ-氨基酸杂合体35~37(图7)的体外抗CQS D6和CQR W2恶性疟原虫活性,发现绝大多数杂合体的IC50在微摩尔级。SAR研究结果表明,氨基酸的构型及AQ C-2位的取代基与活性息息相关,且L-型氨基酸优于D-型。向杂合体36的R1位引入叔丁基对活性有利,但向37的R1位引入叔丁基则对活性不利。进一步研究表明,向这类杂合体引入烷氧基如杂合体38也具有优秀的活性,且长烷氧基链优于短烷氧基。其中,杂合体37a(IC50:258.9和517.9nmol/L)的抗CQS D6和CQR W2恶性疟原虫活性是PQ(IC50:7,722和10810nmol/L)的29.8和20.8倍。在感染伯氏鼠疟的小鼠模型中,杂合体36a和37a(每日4·50mg/kg, 口服给药)可完全治愈(6/6治愈, 治愈率100%)感染小鼠,值得进一步研究。
Sinha等研究发现,绝大多数7-氯喹啉-氨基酸杂合体具有潜在的抗CQS D6和CQR K1恶性疟原虫活性,但大多数杂合体的RI>5。代表物39a,b(IC50:11.51~71.16nmol/L)对所测CQS D6和CQR K1恶性疟原虫具有极高的活性,抗CQR K1恶性疟原虫分别是CQ(IC50:255nmol/L)的3.5和4.6倍。某些7-氯喹啉-抑胃酶氨酸杂合体也具有良好的抗CQS D10和CQR W2恶性疟原虫活性,其中杂合体40(IC50:64nmol/L)的活性最高,其抗CQR W2恶性疟原虫活性是CQ(IC50:412nmol/L)的6.4倍。
(2R,3S)-N-苯甲酰基-3-苯基异丝氨酸-喹啉杂合体41(IC50:130~1000nmol/L)具有潜在的抗CQR W2和耐多药K1恶性疟原虫活性,但弱于CQ(IC50:50和340nmol/L)。SAR显示,含有酯基的杂合体41a~d (IC50:130~390nmol/L)活性优于相应的酰胺衍生物41e,f(IC50:360~1000nmol/L),但向苯基异丝氨酸和喹啉之间引入1,2,3-三氮唑连接子对活性不利,如杂合体42的IC50>1000nmol/L。
2.3 喹啉-唑杂合体
三氮唑、四氮唑、咪唑、吡唑和噻唑具有与各种靶点形成氢键、范德华力等多种非共价键能力,其衍生物具有抗疟疾在内的多种生物活性。显然,将唑与喹啉杂合是获得新型抗疟疾药物的潜在途径。
2.3.1 喹啉-三氮唑杂合体
喹啉-三氮唑杂合体43(图8,IC50:8.9~5330nmol/L)具有中等到优秀的抗CQS NF54和CQR Dd2恶性疟原虫活性,且其中的某些化合物活性高于CQ(IC50:12.1和245nmol/L)。大多数杂合体的RI在1左右,远低于CQ(RI:20.2),提示这类杂合体具有治疗耐药疟疾的潜力。R位含有苄胺的杂合体活性优于相应的苯胺衍生物,且1,2,3-三氮唑与烷胺之间的连接子越短越有利于活性。进一步研究显示,用酰胺取代R位的胺基或向1,2,3-三氮唑引入苯环如杂合体44和45将导致活性的大幅降低。其中,杂合体44a~c(抗CQS NF54和CQR Dd2恶性疟原虫的IC50分别为9.8~15.0nmol/L和50.8~53.5nmol/L)的抗CQS NF54与CQ相当,而抗CQR Dd2活性则是CQ的约5倍,值得进一步研究。

图6 喹啉二聚体25~34的化学结构

图7 喹啉-氨基酸杂合体35~42的化学结构
喹啉-三氮唑杂合体46(IC50:38.75~10.14nmol/L)的抗无性血内期CQS 3D7恶性疟原虫活性弱于CQ(IC50:5.8nmol/L),但某些化合物如46a,46b和46e(IC50:75.01, 2.94和16.01nmol/L)的抗无性血内期CQR RKL-9恶性疟原虫活性优于CQ(IC50:114.4nmol/L),且对有性繁殖CQR RKL-9恶性疟原虫也具有良好的活性,IC50分别为10.71、8.50和12.03μmol/L。SAR研究结果表明,延长连接子的长度对活性不利,但向R位引入卤素如氯和溴则可提高活性。杂合体46b (IC50:40.00, 2.94nmol/L和8.50μmol/L)对所测的所有恶性疟原虫株均显示出良好的活性,值得进一步开发。而无连接子的喹啉-1,2,3-三氮唑杂合体47的抗CQR恶性疟原虫活性在微摩尔级,弱于CQ(纳摩尔级),提示活性与连接子息息相关。
喹啉-1,2,4-三氮唑杂合体48(图8)的抗CQR W2恶性疟原虫SAR显示,与无取代的化合物相比,向R位引入供电子的甲基可提高活性,而吸电子的三氟甲基则对活性不利。代表物48b的抗CQR W2恶性疟原虫IC50为83nmol/L,活性约为CQ(IC50:250nmol/L)的5倍。在感染伯氏鼠疟的小鼠模型中,给药48b,d5时,小鼠寄生虫血症降低66%,值得深入研究。
2.3.2 喹啉-四氮唑杂合体
四氮唑是羧基的生物电子等排体,可替代分子中的羧基以提高脂溶性、改善生物利用度和降低毒副作用。因此,向喹啉核引入四氮唑结构单元是开发新型抗疟疾药物的有效途径之一。
喹啉-四氮唑杂合体49(图9, IC50:6~2979nmol/L)和50(IC50:12~3505nmol/L)具有潜在的抗CQS 3D7, CQR K1和W2恶性疟原虫活性,其中的某些化合物抗CQR K1和W2恶性疟原虫活性甚至优于对照药AQ (IC50:27和401nmol/L)和CQ(IC50:36和59nmol/L),但抗CQS 3D7恶性疟原虫活性均弱于AQ(IC50:1nmol/L)和CQ(IC50:5nmol/L)。对杂合体49而言,叔丁基和二乙胺基分别为R1和R2位最优取代基。抗恶性疟原虫活性顺序为杂合体50a(IC50:12~40nmol/L)>49b(IC50:52~227nmol/L)>>50b(IC50:3385~3505nmol/L),提示连接子的链接方式与活性息息相关,且对位最优。代表物50a(IC50:12和40nmol/L)抗CQR K1和W2恶性疟原虫活性是AQ(IC50:27和401nmol/L)和CQ(IC50:36和59nmol/L)的1.5~10.2倍,值得进一步研究。
进一步研究发现,向R2位引入芳环代替胺基如杂合体51,将会导致杂合体抗CQS 3D7和CQR K1恶性疟原虫活性的大幅降低,但用环己基代替R1位的叔丁基则对活性影响不大。杂合体52的抗恶性疟原虫活性在微摩尔级,远逊于49~51,提示喹啉与四氮唑之间的芳胺连接子对高活性至关重要。杂合体51a(IC50:28.00和77.79nmol/L)和51b(IC50:92.66和73.70nmol/L)具有良好的抗CQS 3D7和CQR K1恶性疟原虫活性,二者抗CQR K1恶性疟原虫活性是CQ (IC50:254nmol/L)的3倍。在感染CQR N-67约氏疟原虫的小鼠模型中,杂合体51c(50mg/kg×d4,腹腔注射给药)在给药d4可抑制99.99%寄生虫血症,且在第28d存活率为60%,治愈率为60%。杂合体51d(50mg/kg×d4,腹腔注射)在给药d4也可抑制99.99%寄生虫血症,且在d28存活率为80%,但治愈率仅为40%。口服给药(100mg/kg×4d)时,二者均可抑制99.99%寄生虫血症,且在d28存活率均为60%。显然,二者极具进一步研究的潜力。
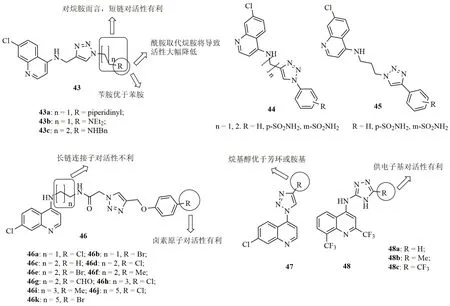
图8 喹啉-三氮唑杂合体43~48的化学结构
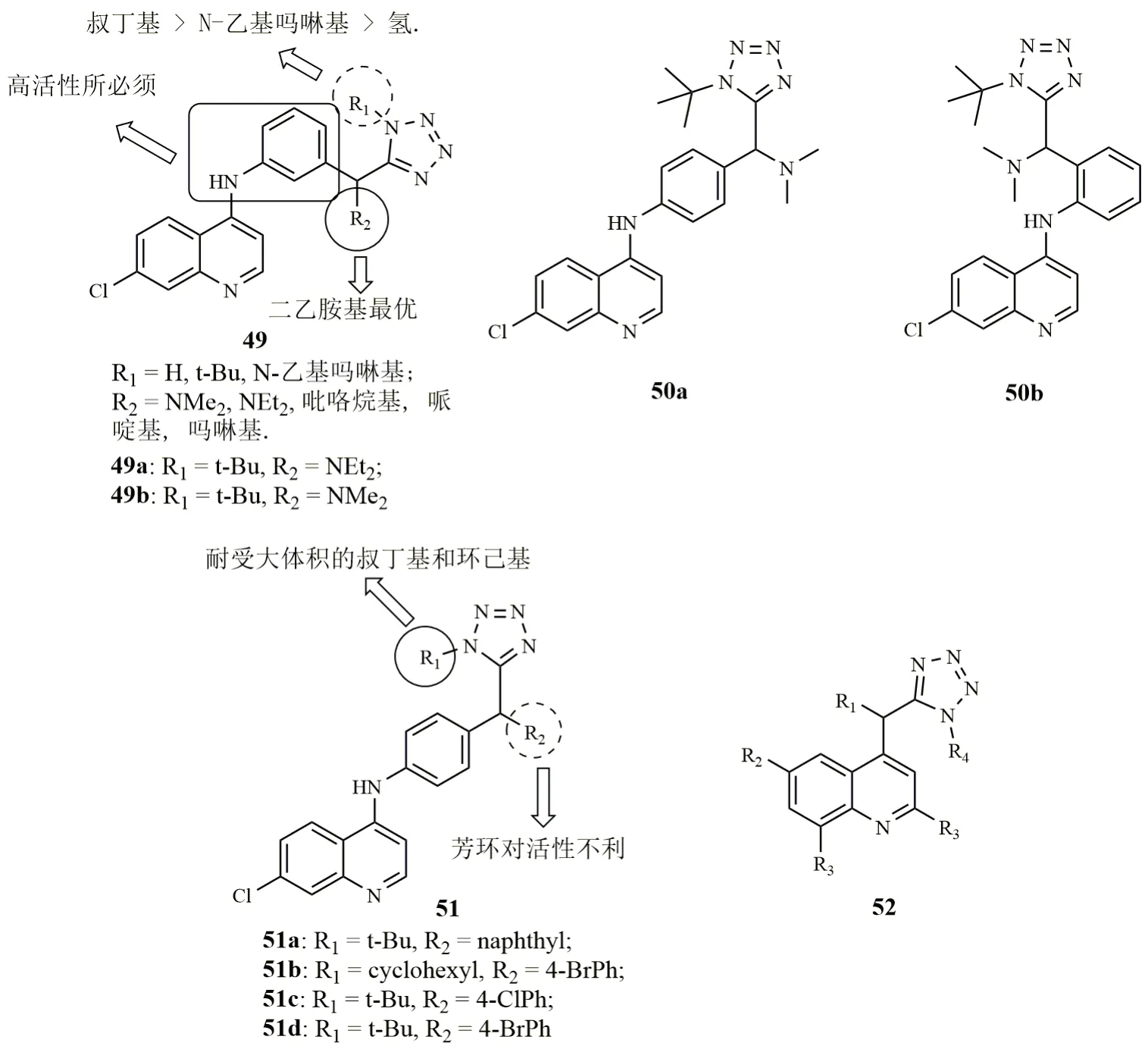
图9 喹啉-四氮唑杂合体49~52的化学结构
2.3.3 喹啉-咪唑杂合体
喹啉-咪唑杂合体53(图10)抗CQS 3D7和CQR K1恶性疟原虫SAR研究结果表明,向R位引入烷基比芳基对活性有利。其中,杂合体53a~e抗CQS 3D7和CQR K1恶性疟原虫的IC50为纳摩尔级,但弱于CQ (IC50:5和255nmol/L),仅杂合体53a(IC50:290nmol/L)的抗CQR K1恶性疟原虫活性与CQ相当。作用机制研究发现,这类杂合体可与血红素结合且可抑制β-血红素的形成,进而诱导恶性疟原虫凋亡。杂合体54a(IC50:148nmol/L)和54n(IC50:66.9nmol/L)具有良好的抗CQR W2恶性疟原虫活性,且后者优于前者,提示向苯环上引入吸电子的氟原子对活性有利。
大多数喹啉-硝基咪唑杂合体55和喹啉-硝基咪唑-四氮唑杂合体56的抗CQR K1恶性疟原虫IC50在纳摩尔级,其中杂合体56(IC50:100~2042nmol/L)的活性普遍优于55(IC50:94.3~11063nmol/L),提示四氮唑的引入可在一定程度上提高活性。对杂合体55而言,化合物55c,d(IC50:821和94.3nmol/L)的活性高于衍生物55a(IC50:11063nmol/L),55b(IC50:4250nmol/L)和55e(IC50:10520nmol/L),提示碱性胺基连接子为高活性所必需。其中,杂合体55d(IC50:94.3nmol/L)的抗CQR K1恶性疟原虫活性是CQ和PQ(IC50:213和643nmol/L)的>2倍,且对L6细胞的毒性(IC50:17.6μmol/L)较低,SI为186.6。喹啉-苯并咪唑杂合体57对VERO细胞的毒性(CC50:>80μmol/L)也较低,且具有潜在的抗CQR W2恶性疟原虫活性(IC50:800nmol/L),但其活性弱于AQ(IC50:50nmol/L)。
2.4 喹啉-查耳酮杂合体
查尔酮也是药物分子常见结构单元,其衍生物具有包括抗疟疾在内的广泛生物活性。某些查尔酮衍生物已被用于治疗包括癌症和心血管疾病在内的多种疾病,故有必要将喹啉与查尔酮杂合。

图10 喹啉-咪唑杂合体53~57的化学结构
1,2,3-三氮唑连接的喹啉-查尔酮杂合体58(图11,IC50:40~5000nmol/L)及其位置异构体59(IC50:300~11700nmol/L)具有潜在的抗CQS D10, CQR D2和W2恶性疟原虫活性,但均弱于CQ(IC50:17,97和69nmol/L)。SAR研究结果表明,杂合体的抗恶性疟原虫活性与R位取代基息息相关,且2,4-二甲氧基(58b,IC50:40,70和90nmol/L)和2,3,4-三甲氧基(59b, IC50:300,300和500nmol/L)较优。其中,杂合体58b (IC50:40,70和90nmol/L)对所测3株恶性疟原虫的活性均较高,且可与CQ(IC50:17,97和69nmol/L)相媲美,但该杂合体水溶性较差导致口服生物利用度较低。进一步研究显示,向喹啉-1,2,3-三氮唑-查尔酮杂合体引入二茂铁并不能进一步提高此类杂合体的抗CQR恶性疟原虫活性。哌嗪连接的喹啉-查尔酮杂合体60(IC50:300~600nmol/L)也具有优秀的抗CQS D10, CQR D2和W2恶性疟原虫活性,且即使在浓度高达100μmol/L时对CHO细胞仍未显示出明显毒性。哌嗪连接的喹啉-查尔酮杂合体61(抗W2恶性疟原虫IC50:410~1840nmol/L)也有类似的结果。进一步研究显示,抑制血红素的形成是这类杂合体的主要作用机制。
喹啉-查尔酮杂合体62(IC50:3.36~42.86nmol/L)的抗CQS3D7恶性疟原虫活性高于63(IC50:80.01~379.82nmol/L),提示喹啉和查尔酮药效团之间的连接子对活性至关重要。其中,杂合体62a~g (IC50:3.36~9.75nmol/L)不仅具有极高的体外活性,在感染CQR N-67约氏疟原虫的小鼠模型中,这些杂合体[100mg/(kg·d), 口服给药]在给药d4可抑制≥99.5%的寄生虫血症。代表物62f,g在给药4d可抑制99.9%的寄生虫血症,略优于CQ[20mg/(kg·d)],但未观测到治愈效果。
喹啉-查尔酮杂合体64(IC50:50~1780nmol/L)的抗CQS D10和CQR D2恶性疟原虫SAR研究结果显示,这类杂合体的活性与二胺基烷基连接子的长度呈正比,即链越长活性越高。其中,杂合体64e(IC50:50和70nmol/L)的抗CQS D10恶性疟原虫与CQ(IC50:50nmol/L)相当,抗CQR D2恶性疟原虫活性则是CQ(IC50:120nmol/L)的1.7倍。喹啉-查尔酮杂合体65(IC50:69.11~255.4nmol/L)和66(IC50:47.3~900nmol/L)也具有潜在的抗CQS 3D7和CQR K1恶性疟原虫活性,但抗CQS 3D7恶性疟原虫活性弱于CQ(IC50:7.68nmol/L)。杂合体65(IC50:134.1~252.7nmol/L)的抗CQR K1恶性疟原虫活性优于66(IC50:229.1~900nmol/L),提示酰胺的引入有利于克服耐药性。其中,杂合体65b(IC50:134.1nmol/L)的抗CQR K1恶性疟原虫活性是CQ(IC50:463nmol/L)的3.4倍,值得进一步研究。
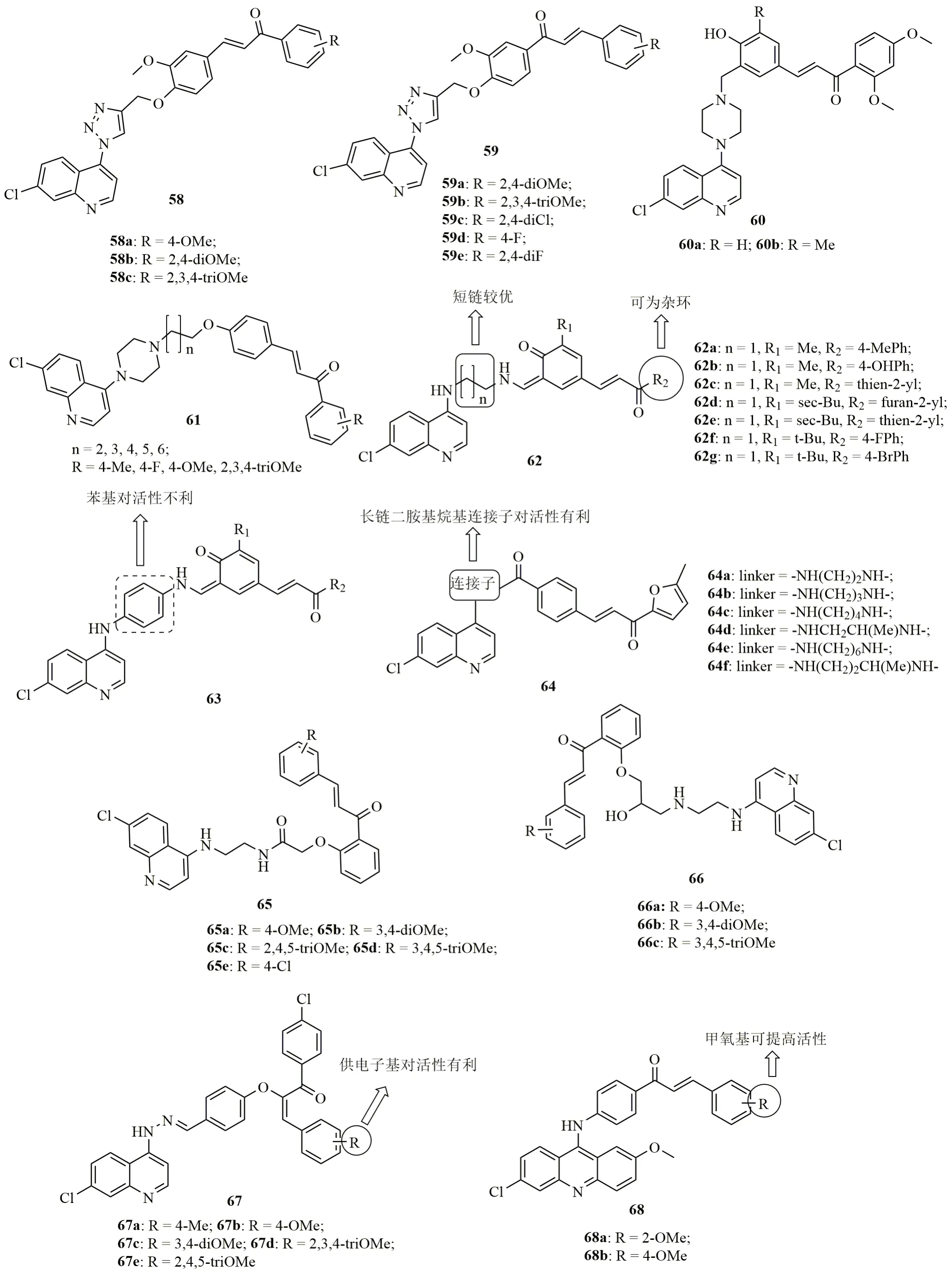
图11 喹啉-查尔酮杂合体58~68的化学结构
喹啉-查尔酮杂合体67(IC50:48.05~314.86nmol/L)具有较高的抗 CQS 3D7和CQR K1恶性疟原虫活性,且抗CQR K1恶性疟原虫活性高于CQ(IC50:463nmol/L),但抗CQS 3D7恶性疟原虫活性弱于CQ(IC50:7.68 nmol/L)。向苯环上引入甲基和甲氧基等供电子基可提高活性,而吸电子基如氟、氯、溴和硝基则对活性不利。甲氧基的数量对活性有一定影响,且个数越多活性越高。代表物67a~e(IC50:82.93~97.87nmol/L)的IC50<100nmol/L,可作为先导物进一步优化。
吖啶-查尔酮杂合体68(抗CQS 3D7和CQR Dd2恶性疟原虫的IC50分别为300~4800nmol/L和150~5000nmol/L)的抗CQS 3D7恶性疟原虫活性明显弱于CQ(IC50:40和170nmol/L),但某些化合物的抗CQR Dd2恶性疟原虫活性略优于CQ。SAR显示,甲氧基对活性有利,且杂合体68a(IC50:150nmol/L)的抗CQR Dd2恶性疟原虫活性略优于CQ。
2.5 喹啉-肉桂酸杂合体
喹啉-肉桂酸杂合体69(图12,IC50:>10000nmol/L)未显示出任何抗CQR W2恶性疟原虫活性,但其二肽连接的衍生物70(IC50:830~10800nmol/L)则具有弱到中等强度的活性,提示连接子与活性息息相关。对杂合体70而言,向苯环引入硝基或对位引入烷基对活性有利。其中,杂合体70b(IC50:830nmol/L)的抗CQR W2恶性疟原虫活性最高,但弱于ART和CQ(IC50:8.76和76nmol/L)。进一步研究发现,用胺基-酰胺或胺基-酯替代二肽可大幅提高活性,如杂合体71对血内期(CQS 3D7和CQR W2恶性疟原虫的IC50为11.0~121.9nmol/L)和肝期(伯氏鼠疟原虫,IC50:1090~6470nmol/L)具有良好的活性,且含有酰胺的杂合体活性高于相应的酯基衍生物。C-7位的氯为高活性所必需,且用PQ核代替CQ核将导致活性的急剧降低,提示喹啉核对活性有显著影响。延长连接子的碳链长度或向苯环的对位引入烷基或卤素对活性有利,但用烷基取代芳基对活性不利。其中,杂合体71a(IC50:15.63和11.0nmol/L)的CQS 3D7和CQR W2恶性疟原虫活性与ART(IC50:23.5和9.5nmol/L)相当,而是CQ(IC50:52.45和138nmol/L)的3.3和12.5倍。值得一提的是,与CQ(IC50:15900nmol/L)相比,该杂合体(IC50:2500nmol/L)抗伯氏鼠疟原虫活性也有所增强,值得进一步研究。
吖啶-肉桂酸杂合体72(抗CQR W2恶性疟原虫IC50:126~892nmol/L)和73(IC50:3.2~131.0nmol/L)也具有良好的抗CQS 3D7,CQR W2和Dd2恶性疟原虫及伯氏鼠疟原虫活性,且杂合体73的活性高于相应的衍生物72,提示吖啶上的取代基与活性息息相关。对杂合体73而言,供电子的烷基如甲基和异丙基对抗CQS 3D7,CQR W2和Dd2恶性疟原虫活性的贡献优于吸电子的氟和溴等。代表物73e(抗CQS 3D7, CQR W2和Dd2恶性疟原虫及伯氏鼠疟原虫的IC50分别为8.9~28.9nmol/L和1800nmol/L)的抗CQS 3D7恶性疟原虫活性与CQ(IC50:21.0nmol/L)相当,抗CQR W2和Dd2恶性疟原虫及伯氏鼠疟原虫则是CQ(抗CQR W2和Dd2恶性疟原虫及伯氏鼠疟原虫的IC50分别为IC50:107.5, 225.8nmol/L和>15000nmol/L)的3.7~>8.3倍,可作为先导物进一步优化。
2.6 喹啉-二茂铁杂合体
二茂铁具有三明治夹心结构,其衍生物具有抗菌和抗疟疾等多种生物活性。值得一提的是,喹啉-二茂铁杂合体二茂铁喹(FQ)对CQS和CQR恶性疟原虫均具有优秀的活性,且无免疫毒性。进一步研究发现,二茂铁核可克服PfCRT,进而使喹啉在食物泡累积。显然,将喹啉与二茂铁杂合是克服CQ耐药性的有效途径。
喹啉-二茂铁杂合体74(图13, IC50:4.9~16.5nmol/L)的抗CQS NF54,CQR K1和Dd2恶性疟原虫活性可与FQ(IC50:1.6~20.0nmol/L)相媲美,抗CQR K1和Dd2恶性疟原虫活性则是CQ(IC50:316.3和527.0nmol/L)的19.1和107.5倍。SAR研究结果表明,二茂铁核对抗CQR恶性疟原虫至关重要,移除该结构单元将导致活性大幅降低。
喹啉-二茂铁杂合体75的抗CQS D10和CQR Dd2恶性疟原虫SAR显示,喹啉和二茂铁之间的连接子及二茂铁与酰胺之间的碳链长度与活性息息相关,且烷基二胺基为最佳连接子。代表物75c的抗CQR Dd2恶性疟原虫IC50为80nmol/L,活性优于PQ(IC50:820nmol/L)和CQ(IC50:120nmol/L)。进一步研究发现,喹啉和二茂铁之间的酰胺可被酯基取代,且对含有酯基的杂合体76而言,支链烷基连接子优于直链烷基连接子。杂合体76a~d(IC50:130~660nmol/L)的抗CQR Dd2恶性疟原虫IC50在纳摩尔级别,且代表物76d(IC50:130nmol/L)的活性是CQ(IC50:340nmol/L)的2.6倍。显然,杂合体75c和76d值得进一步开发。
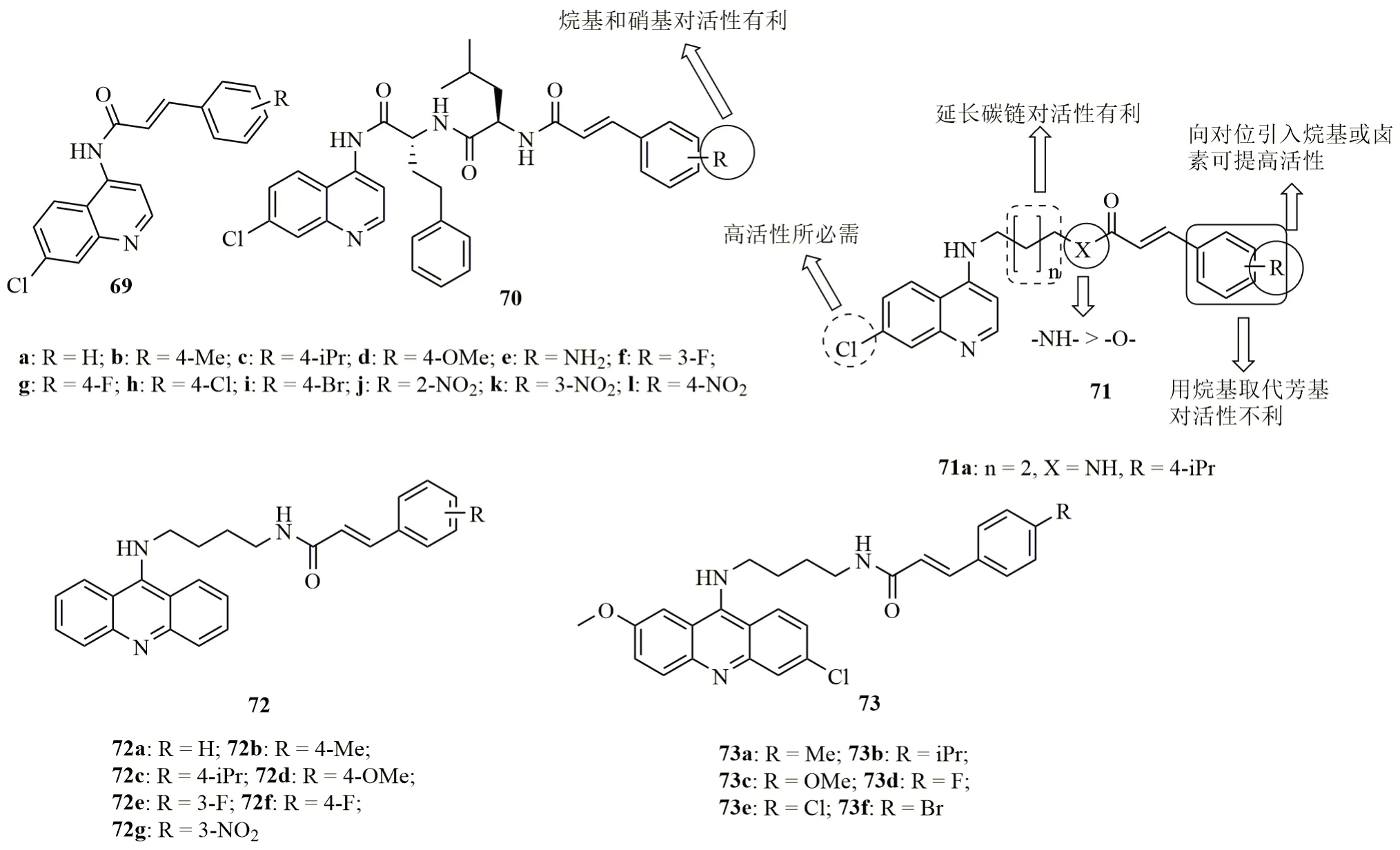
图12 喹啉-肉桂酸杂合体69~73的化学结构
喹啉-二茂铁杂合体77(IC50:16.1~1136nmol/L)的抗CQS D10, CQR Dd2和K1恶性疟原虫活性普遍高于杂合体78(IC50:91.3~669.0nmol/L),提示两个药效团之间的连接方式对活性有显著影响。绝大多数杂合体的RI<1,提示这类杂合体可能具有全新的作用机制。杂合体77b(IC50:16.1nmol/L)和77d(IC50:23.1nmol/L)对CQR K1和Dd2的活性最高,且活性可与FQ(IC50:14.0和19.0nmol/L)媲美,而是CQ (IC50:758.0和180.3nmol/L)的47.0和7.8倍。进一步研究发现,向喹啉-二茂铁杂合体引入糖结构单元如化合物79(IC50:47.7~242nmol/L)也具有良好的抗CQS D10, CQR Dd2和K1恶性疟原虫活性,但弱于相应的无糖衍生物。
刚性的连接子如哌嗪和甲基哌嗪连接的喹啉-二茂铁杂合体80对抗CQS D10和CQR Dd2恶性疟原虫活性不利,但烷基二胺基则可提高活性。杂合体80d抗CQS D10和CQR Dd2恶性疟原虫的IC50分别为40和10nmol/L,活性优于CQ(IC50:50和150nmol/L)。值得一提的是,所有杂合体的RI均在0.1~0.4之间,提示这类杂合体具有治疗耐药疟疾的潜力。
杂合体81(IC50:6.90~35.57nmol/L)的抗恶性疟原虫SAR研究结果显示,X位为硅原子时比碳原子对抗 CQS NF54恶性疟原虫更为有利,但对抗CQR Dd2恶性疟原虫活性影响不大。代表物81b(IC50:6.90和13.87nmol/L)的抗CQR Dd2恶性疟原虫活性与ART (IC50:16.91nmol/L)相当,但抗 CQS NF54和CQR Dd2恶性疟原虫活性高于FQ(IC50:24.90和17.34nmol/L)和CQ(IC50:13.66和301.37nmol/L)。此外,该杂合体81b(IC50:15.36μmol/L)对CHO细胞的毒性较低,SI >1107,可作为先导物进一步研究。
喹啉-二茂铁杂合体82(IC50:13.8~39.3μmol/L)的抗CQS D10和CQR W2恶性疟原虫较弱,提示喹啉与二茂铁之间的连接子对活性至关重要。
2.7 喹啉-吲哚/吡咯杂合体
吡咯和吲哚包括靛红广泛存在于自然界中,其衍生物具有包括抗疟疾在内的多种药理活性。因此,将吡咯和吲哚与喹啉杂合也是目前开发新型抗疟疾药物的常用手段。
研究发现,组蛋白去乙酰化酶(HDAC)抑制剂对药敏型和耐药型恶性疟原虫均显示出潜在的活性,值得进一步研究。喹啉-靛红HDAC抑制剂83(图14)对所测的24株临床分离耐多药恶性疟原虫和25株临床分离间日疟原虫具有良好的活性,其平均IC50分别为266和278nmol/L,但弱于对照药CQ、AQ、PQ, MQ和ART(平均IC50:0.74~40.7nmol/L)。
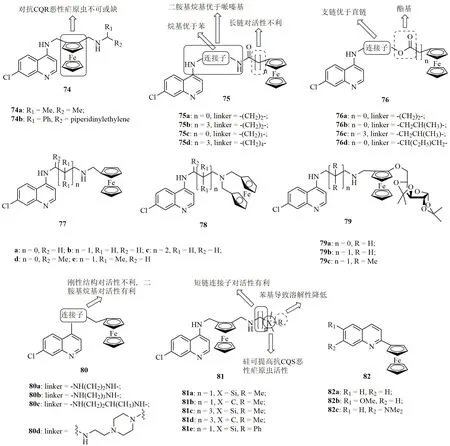
图13 喹啉-二茂铁杂合体74~82的化学结构
对包含喹啉-吲哚杂合体84在内的一系列喹啉衍生物的抗CQS 3D7和CQR K1恶性疟原虫活性评价结果表明,化合物84a(IC50:16.88和122.7nmol/L)的活性优于84b(IC50:88.86和259.26nmol/L),提示向喹啉母核C-7引入氯比引入三氟甲基对活性更有利。尽管二者的抗CQS 3D7恶性疟原虫活性弱于CQ(IC50:5.46nmol/L),但杂合体84a的抗CQR K1恶性疟原虫活性高于CQ(IC50:255nmol/L)。向喹啉的C-8位引入羟基将导致活性的大幅降低,如杂合体85在浓度高达5μmol/L时对CQR Dd2恶性疟原虫的生长抑制率仅为<10%。
喹啉-吲哚杂合体86(IC50:100~2200nmol/L)及其盐87(IC50:120~1600nmol/L)具有潜在的抗CQS 3D7和CQR K1恶性疟原虫活性,且对杂合体86而言,双键对抗CQR K1恶性疟原虫至关重要,如杂合体86a,b (IC50:390和330nmol/L)的活性远优于单键衍生物86c (IC50:2200nmol/L)。对杂合体87而言,用丁基取代R1位的甲基对活性不利。有趣的是,与其它喹啉杂合体不同,向这类杂合体的R2位引入氯原子不仅不会提高活性,反而对活性不利。向R3或R4位引入甲基对抗CQS 3D7和CQR K1恶性疟原虫活性均不利。代表物87b(IC50:120nmol/L)的抗CQR K1恶性疟原虫活性高于CQ(IC50:300nmol/L),可作为先导物进一步优化。

图14 喹啉-吲哚杂合体83~87的化学结构
吲哚[3,2-b]喹啉88(图15)的抗CQS 3D7和CQR W2恶性疟原虫SAR研究结果表明,向R1和R2位引入氯原子可提高活性,且R3位为直链烷胺基优于环烷胺基。杂合体的RI为0.3~0.7,可能具有全新的作用机制。代表物88a(IC50:25nmol/L)的抗CQR W2恶性疟原虫活性与ART(IC50:14nmol/L)相当,而是CQ (IC50:14nmol/L)的2.6倍。进一步研究发现,醚基可被胺基取代,且对杂合体89而言,与相应的无取代化合物相比,向R1位引入氯对活性不利。杂合体89a (IC50:20nmol/L)的抗CQR W2恶性疟原虫活性最高,是CQ (IC50:229nmol/L)的11.4倍。
白叶藤碱衍生物90具有优秀的抗CQS 3D7, CQR W2, V1/S(耐CQ和乙胺嘧啶)和D6(CQ敏感, 但耐MQ)恶性疟原虫活性,IC50<100nmol/L,且RI为1。对R1和R2位而言,氯原子的引入对活性影响不大,但会增大对VERO细胞的毒性。对R3位而言,与相应的末端伯胺相比,仲胺和季铵盐对抗CQS 3D7和CQR D6恶性疟原虫更为有利。与相应的直链烷基连接子相比,支链连接子会导致活性大幅下降。代表物90c (IC50:20~32nmol/L)的抗CQR D6恶性疟原虫活性略弱于CQ(IC50:14.2~138nmol/L),但抗CQR W2和V1/S恶性疟原虫活性则是CQ的>4倍。不仅如此,该杂合体 (IC50:2.5μmol/L)对VERO细胞的毒性较低,SI为78,值得深入研究。
异白叶藤碱衍生物91的抗CQS 3D7, CQR K1和耐多药SKF58及SRIV35(同时耐CQ和MQ)恶性疟原虫具有优秀的活性,IC50在纳摩尔级。SAR显示,向喹啉环引入氟原子可提高活性,如化合物91a(IC50:37.9~92.4nmol/L)对CQS 3D7, CQR K1和耐多药SKF58及SRIV35恶性疟原虫的活性极高,抗耐多药SKF58及SRIV35恶性疟原虫活性不亚于CQ(IC50:76和145nmol/L),QN(IC50:199和431nmol/L)和MQ (IC50:55和65nmol/L)。
吲哚[2,3-b]喹啉衍生物92(抗CQS NF54和CQR K1恶性疟原虫的IC50分别为2.16~24.8nmol/L和11.9~30.3nmol/L)和93(抗CQS NF54和CQR K1恶性疟原虫的IC50分别为1.18~16.7nmol/L和16.1~68.4nmol/L)的抗CQS NF54恶性疟原虫活性与CQ(抗CQS NF54和CQR K1恶性疟原虫的IC50分别为9.4和210nmol/L)相当,而抗CQR K1恶性疟原虫活性则是CQ的3.0~17.6倍。SAR研究表明,用脲或硫脲(93)代替胺基(92)可提高抗CQS NF54恶性疟原虫活性,但会降低抗CQR K1恶性疟原虫活性。对杂合体93而言,含有硫脲结构单元的化合物抗CQS NF54和CQR K1恶性疟原虫活性优于相应的含脲衍生物。在感染伯氏鼠疟原虫的雌性小鼠模型中,杂合体92a,b(20mg/kg, 腹腔注射给药)可在d4,7降低33.4%和72.5%的寄生虫血症。
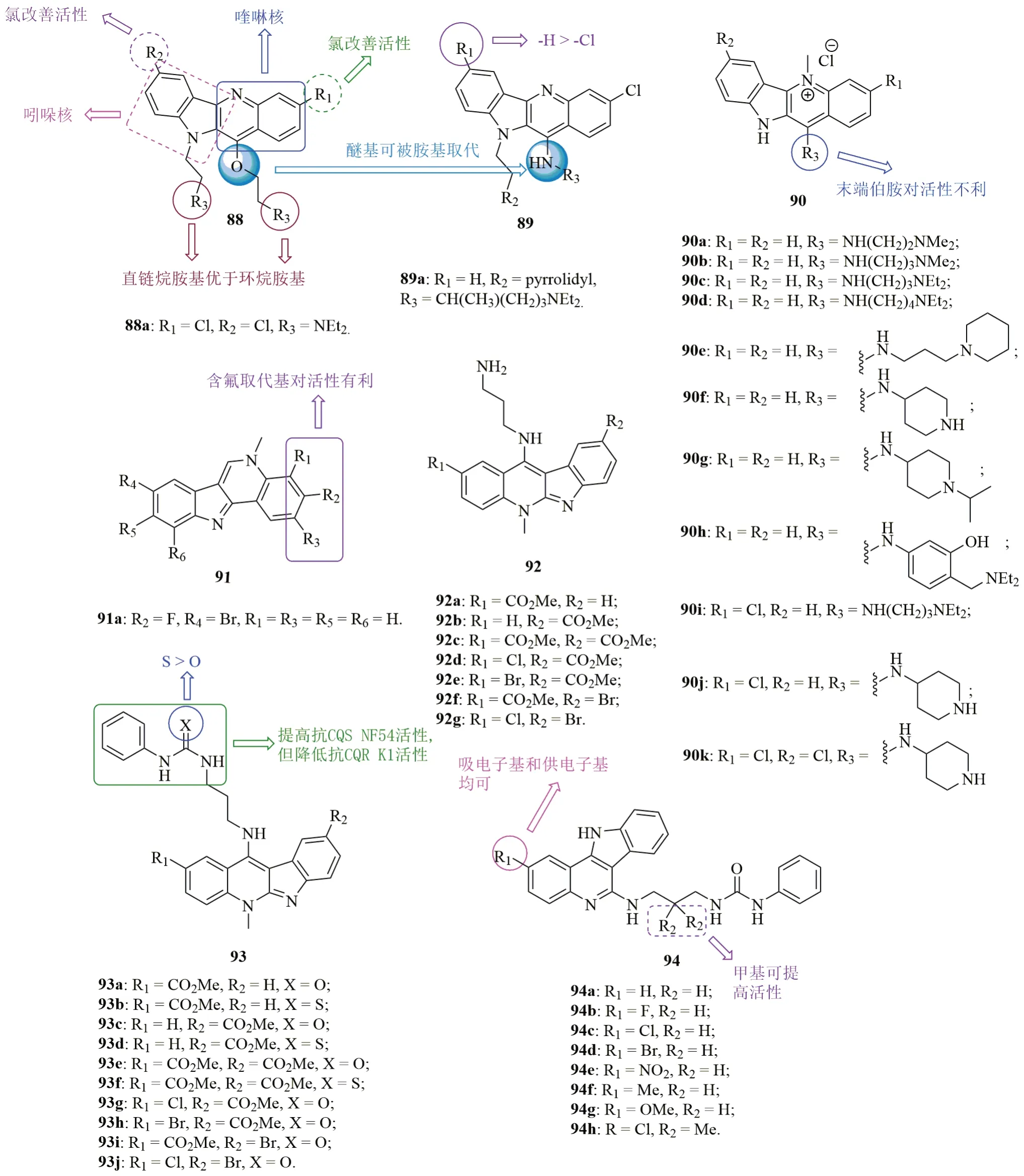
图15 化合物88~94的化学结构
吲哚[3,2-c]喹啉衍生物94抗CQS NF54和CQR K1恶性疟原虫的IC50分别为10.6~27.0nmol/L和16.9~53.7nmol/L,活性与CQ(IC50:9.4和209.5nmol/L)相当或更优。SAR显示,与无取代衍生物相比,向R1位引入吸电子基或供电子基和向R2位引入甲基均可提高活性。代表物94h(IC50:10.6nmol/L)的抗CQS NF54恶性疟原虫活性略低于CQ和ART(IC50:9.4和4.3nmol/L),但其(IC50:16.9nmol/L)抗CQR K1恶性疟原虫活性则是CQ的12.3倍。
1,2,3-三氮唑连接的喹啉-靛红杂合体95(图16, IC50:118~346nmol/L)及其还原产物96(IC50:69.0~509nmol/L)具有潜在的抗CQR W2恶性疟原虫活性,但活性弱于CQ(IC50:60.0nmol/L)和ART(IC50:7.00nmol/L)。7-氯喹啉母核对活性至关重要,无该结构片段的杂合体无活性。靛红和1,2,3-三氮唑之间的连接子碳个数与活性息息相关,且奇数个碳优于偶数个碳。对杂合体95而言,靛红C-5位取代基对活性影响较小,但对杂合体96而言,向该位点引入氯和甲基对活性不利。其中,杂合体96c(IC50:69.0nmol/L)的抗CQR W2恶性疟原虫活性与CQ (IC50:60.0nmol/L)相当。用哌嗪-烷基(97, IC50:270~7520nmol/L)或哌嗪-炔基(98, IC50:420~1120nmol/L)取代1,2,3-三氮唑做连接子会降低活性,而β-胺基-醇连接的杂合体99(IC50:11.7~488.1nmol/L)则显示出更高的活性。对杂合体99而言,向靛红的C-5位引入氯原子可提高活性,且偶数碳原子链优于奇数链。进一步研究发现,用烷基二胺基(100, IC50:219~1960nmol/L)取代哌嗪基对活性不利。代表物99b(IC50:11.7nmol/L)的抗CQR W2恶性疟原虫活性是CQ(IC50:36.37nmol/L)的3.1倍,且对HCT116细胞(IC50:>20μmol/L)无毒,可作为先导物进一步研究。
喹啉-靛红杂合体101(IC50:140~420nmol/L)的抗CQR W2恶性疟原虫SAR显示,向靛红母核引入氯原子可提高活性,且氯原子所在位置与活性息息相关,顺序为8- > 7- > 6- > 5-。与无取代杂合体101a (IC50:420nmol/L)相比,向靛红的N-1位引入甲基(101b, IC50:290nmol/L)对活性有利。其中,化合物101e(IC50:140nmol/L)的抗CQR W2恶性疟原虫活性可与CQ(IC50:140nmol/L)相媲美。
喹啉-吡咯杂合体102(图17, IC50:400~1410nmol/L)具有潜在的抗CQR W2恶性疟原虫活性,且CQ衍生物102a~c(IC50:400~990nmol/L)活性高于PQ衍生物102d(IC50:1410nmol/L),提示喹啉母核对活性影响较大。对CQ衍生物而言,延长连接子的长度对活性有利,且代表物102c(IC50:400nmol/L)的活性优于CQ和PQ(IC50:590和1890nmol/L)。此外,该杂合体(IC50:187.3μmol/L)对BGM细胞的毒性极低,SI为468。
吡咯[2,3-c]喹啉103抗CQS 3D7和耐多药K1恶性疟原虫的IC50分别为39和41nmol/L,且对HepG2细胞(EC50:>250μmol/L)无毒,SI>6400。化合物103 (IC50:28~50nmol/L)不仅可快速抑制血内期各种形态的恶性疟原虫,也可抑制肝期恶性疟原虫,且其与青蒿琥酯联合使用具有协同作用。在感染伯氏鼠疟原虫的小鼠模型中,该化合物(50mg/kg,口服给药)可在给药d5降低62%的寄生虫血症,且口服给药1000mg/kg时,小鼠依然存活,提示该化合物的安全性良好。
2.8 喹啉-内酰胺杂合体
脲连接的β-内酰胺-7-氯喹啉杂合体104(图18, IC50:42.38~193.15nmol/L)及其草酰胺衍生物105(IC50:34.97~120.65nmol/L)具有良好的抗CQR W2恶性疟原虫活性,且其中的7个化合物活性高于CQ(IC50:59.09nmol/L)。与脲连接的杂合体104相比,草酰胺连接的杂合体105的活性更高,提示连接子与活性息息相关。与R位为环己基的杂合体相比,4-甲基苯基衍生物的活性更高,提示β-内酰胺上的取代基也可影响活性。其中,代表物105d(IC50:34.97nmol/L)的抗CQR W2恶性疟原虫活性最高,且对HeLa细胞(IC50:>10μmol/L)未显示出毒性。
γ-羟基-γ-内酰胺-喹啉杂合体106(IC50:43~800nmol/L)和107(IC50:56~667nmol/L)具有良好的抗CQS 3D7和CQR W2恶性疟原虫活性,且相当一部分杂合体的抗CQR W2恶性疟原虫活性优于CQ(IC50:750nmol/L)。与相应的烷基连接子相比,含有胺基的连接子对活性更优,且喹啉母核C-7位的氯原子对高活性不可或缺。对杂合体106而言,向喹啉的C-3位(R2位)引入三氟乙酰基对活性不利,且R3位为硫醚的杂合体优于相应的砜衍生物。对杂合体107而言,向R1位引入芳环比甲基更有利。其中,8个杂合体抗CQS 3D7和CQR W2恶性疟原虫的IC50<100nmol/L,抗CQS 3D7恶性疟原虫活性与CQ(IC50:30nmol/L)相当,抗CQR W2恶性疟原虫活性是CQ(IC50:750nmol/L)的>7.5倍。进一步研究发现,与相应的游离化合物相比,柠檬酸盐的活性更高,如杂合体106a(IC50:19和42nmol/L)和107a(IC50:26和49nmol/L)柠檬酸盐对CQS 3D7和CQR W2恶性疟原虫的活性极高。
2.9 喹啉-大环内酯杂合体
大环内酯类抗生素具有良好的体内外预防疟疾活性,且这类抗生素与喹啉类抗疟药物联合使用在耐多药疟疾感染的小鼠模型中具有协同作用。基于此,将喹啉与大环内酯杂合可能会获得对耐药甚至耐多药疟疾具有良好疗效的新型药物。
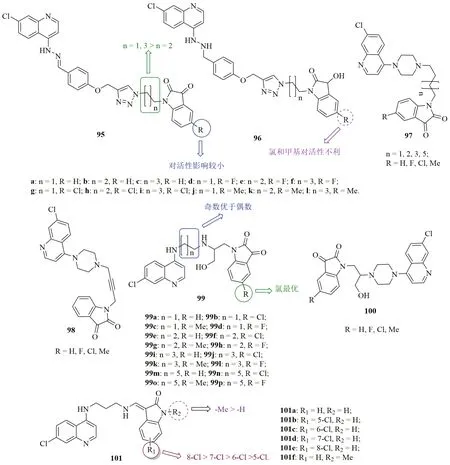
图16 喹啉-靛红杂合体95~101的化学结构
喹啉-阿奇霉素杂合体108(图19, 抗CQS NF54和CQR W2恶性疟原虫的IC50分别为3~636nmol/L和2~295nmol/L)的活性是阿奇霉素(IC50:11,513和2680nmol/L)的>19倍,且所有杂合体抗CQR W2恶性疟原虫均活性高于CQ(IC50:431nmol/L)。SAR显示,与无取代杂合体相比,在R位引入Cladinosyl可明显提高活性。连接子对活性有显著影响,且烷基连接子优于含有酰胺的连接子。代表物108c(IC50:3和2nmol/L)的抗CQS NF54和CQR W2恶性疟原虫是CQ (IC50:16和431nmol/L)的5.3和215.5倍,极具进一步开发前景。
胺基-醚相连的喹啉-阿奇霉素杂合体109(抗CQS NF54和CQR W2恶性疟原虫的IC50分别为14~1657nmol/L 和9~815nmol/L)和110(抗CQS NF54和CQR W2恶性疟原虫的IC50分别为141~1367nmol/L和46~432nmol/L)也显示出潜在的抗恶性疟原虫活性。对杂合体109而言,喹啉母核C-7位的氯原子为高活性所必需。对杂合体110而言,R2位为甲基时对活性无显著影响,但连接位点与活性息息相关,且喹啉-3-基最优。
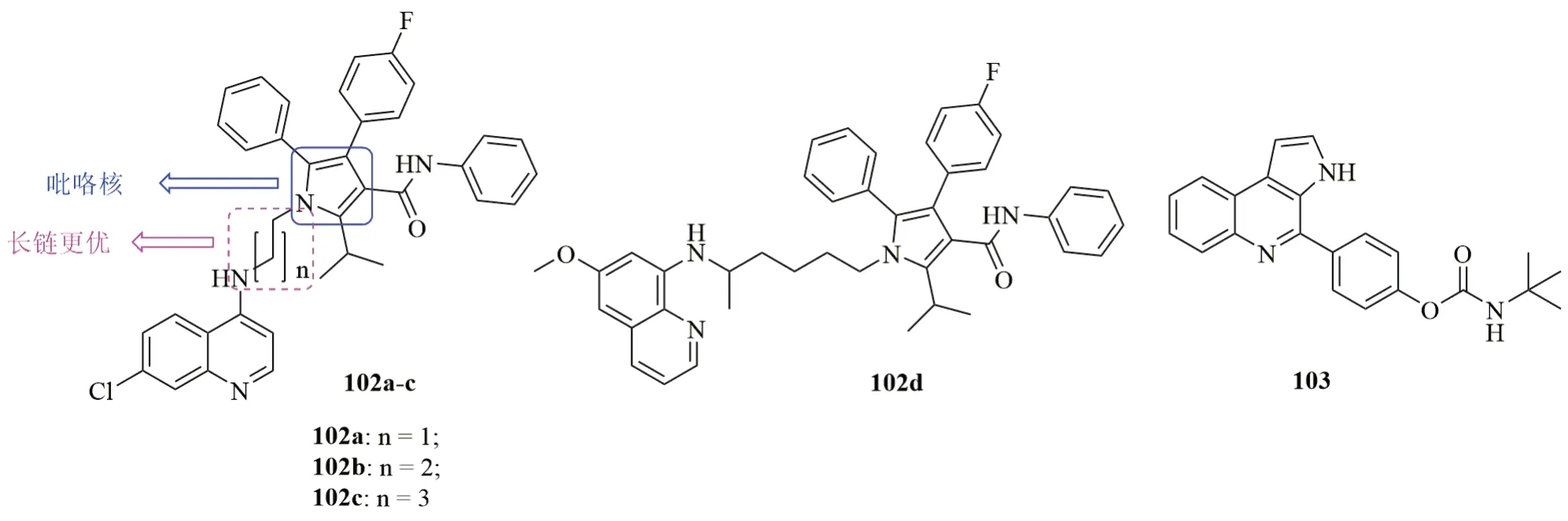
图17 喹啉-吡咯杂合体102和103的化学结构
喹啉-阿奇霉素杂合体111(IC50:13~437nmol/L)的抗耐多药TM91C235恶性疟原虫活性优于相应的衍生物112(IC50:62~1969nmol/L),提示喹啉与胺基之间的亚甲基对活性不利。SAR显示喹啉C-7位的氯原子、R1位的Cladinosyl和R2位的Desosaminyl为高活性所必需的基团。在感染伯氏鼠疟原虫的小鼠模型中,与阿奇霉素组相比,杂合体111c,d(5mg/kg, 静脉注射给药,每日2次,连续给药3d)组小鼠的存活时间分别延长了9和3d。杂合体111c在静脉注射给药15和20mg/kg和腹腔注射给药20mg/kg时,所有小鼠(5/5)均可被治愈,而相同给药方式的阿奇霉素组治愈率分别为2/5, 1/5和4/5,而在最高给药剂量CQ组的治愈率仅为2/5。优良的体内外活性加之良好的药代动力学性质,使得杂合体111c可作为先导物进一步研究。
其它喹啉-大环内酯杂合体如1,2,3-三氮唑连接的喹啉-疏螺体素杂合体113(IC50:0.11和0.096nmol/L)具有良好的抗CQS FCR3和CQR K1恶性疟原虫活性,且活性是疏螺体素(IC50:1.79和1.94nmol/L)的>10倍。此外,杂合体113(IC50:1,506nmol/L)对MRC-5细胞毒性较低,SI>13400。
2.10 喹啉-嘧啶杂合体
嘧啶衍生物是潜在的二氢叶酸还原酶(DHFR)抑制剂,故将喹啉(疟原虫色素抑制剂)与嘧啶(DHFR抑制剂)杂合可能会获得具有双重作用机制的新型抗疟疾候选物。
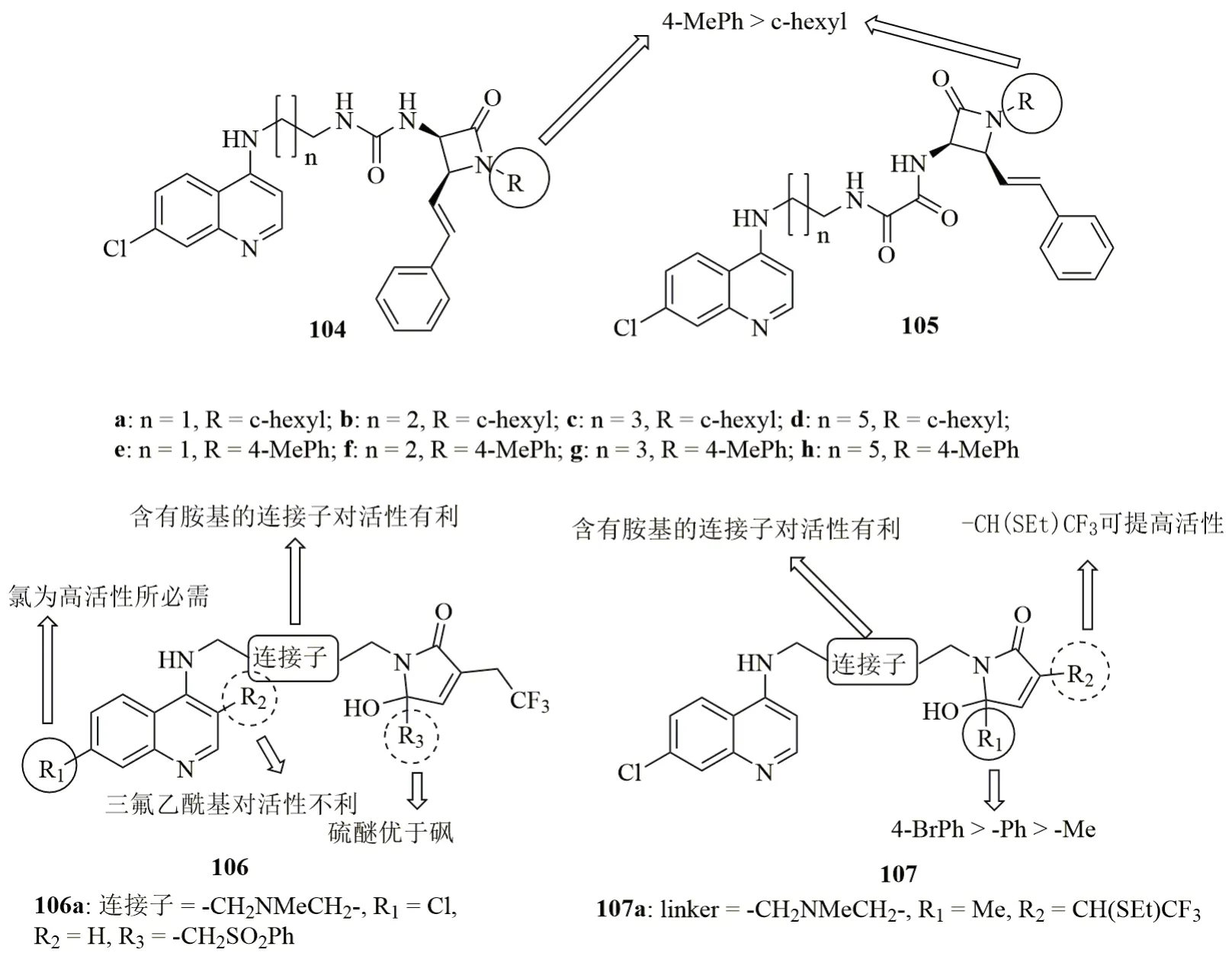
图18 喹啉-内酰胺杂合体104~107的化学结构

图19 喹啉-大环内酯杂合体108~113的化学结构
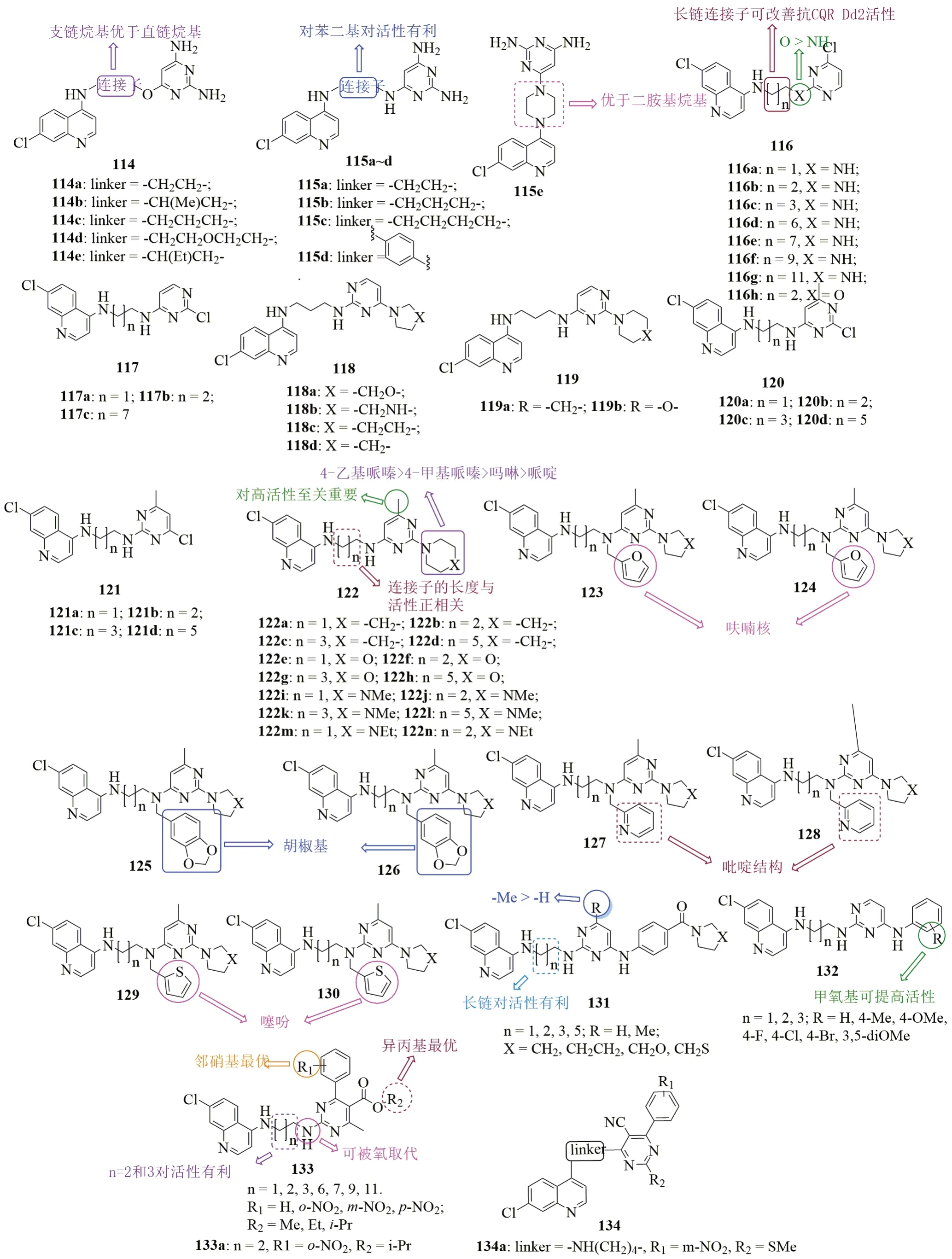
图20 烷基二胺基连接的喹啉-嘧啶杂合体114~134的化学结构
喹啉-嘧啶杂合体114(图20,抗CQS 3D7和CQR W2恶性疟原虫的IC50分别为157~837nmol/L和210~4103nmol/L)和115(抗CQS 3D7和CQR W2恶性疟原虫的IC50分别为70~850nmol/L和107~1263nmol/L)抗恶性疟原虫SAR研究结果显示,醚键连接的杂合体114抗CQS 3D7和CQR W2恶性疟原虫活性优于相应的胺基连接衍生物115,杂合体116(抗CQS 3D7和CQR W2恶性疟原虫的IC50分别为179~3340nmol/L和193~7678nmol/L)也观测到了类似的结果。对杂合体114而言,支链连接子优于直链连接子。对杂合体115而言,与烷基连接子相比,苯基(115d)和哌嗪基(115e)更优。对杂合体116而言,延长连接子有利于抗CQR Dd2恶性疟原虫,但对抗CQS 3D7恶性疟原虫不利。与杂合体116相比,其位置异构体117(IC50:207~883nmol/L)的抗CQR Dd2恶性疟原虫活性更高。用含氮杂环(118和199)代替嘧啶上的氯可在一定程度上提高抗CQR Dd2恶性疟原虫活性更高,但胺基烷基醇则对活性不利。喹啉-嘧啶杂合体120~122(抗CQS 3D7恶性疟原虫的IC50分别为120~440nmol/L, 140~240nmol/L和5~30nmol/L; 抗CQR Dd2恶性疟原虫的IC50分别为500~700nmol/L, 580~1170nmol/L和16-210nmol/L)、含有芳环的杂合体123~130(123和124连有呋喃结构片段, 抗CQS 3D7和CQR Dd2恶性疟原虫的IC50分别为38~61nmol/L和39~257nmol/L; 125和126含有胡椒基, 抗CQS 3D7和CQR Dd2恶性疟原虫的IC50分别为20~150nmol/L和50~990nmol/L; 127和128含有吡啶结构片段,抗CQS 3D7和CQR Dd2恶性疟原虫的IC50分别为30~1193nmol/L和39~1143nmol/L;129和130含有噻吩杂环,抗CQS 3D7和CQR Dd2恶性疟原虫的IC50分别为32.8~83.7nmol/L和18.9~409.1nmol/L)、含有酰胺的杂合体131(抗CQS 3D7和CQR Dd2恶性疟原虫的IC50分别为32~204nmol/L和32~1018nmol/L)和含有芳胺的杂合体132(抗CQS 3D7和CQR Dd2恶性疟原虫的IC50分别为19~6022nmol/L和46~9994nmol/L)也具有类似的SAR。对杂合体122而言,嘧啶核上含氮杂环取代基对活性的贡献顺序为4-乙基哌嗪>4-甲基哌嗪>吗啉>哌啶。连接子的长度与活性正相关,且嘧啶上的甲基对高活性至关重要。杂合体122的抗CQS 3D7恶性疟原虫活性极高,IC50≤30nmol/L,活性优于CQ(IC50:40nmol/L),且不亚于ART(IC50:10nmol/L)。绝大多数杂合体122抗CQR Dd2恶性疟原虫的IC50< 100nmol/L,与ART(IC50:10nmol/L)处于同一水平,而优于CQ(IC50:390nmol/L)。在感染伯氏鼠疟原虫的小鼠模型中,杂合体122i(IC50:5和30nmol/L)和122m(IC50:6和60nmol/L)可在给药剂量为30mg/kg时完全消除寄生虫血症,且二者的治愈率分别为80%和20%,而CQ(100mg/kg)组无小鼠治愈。显然,杂合体122i值得深入开发。
向嘧啶片段引入苯基或酯基也耐受,如杂合体133抗CQS 3D7和CQR K1恶性疟原虫的IC50分别为18.2~1955.1nmol/L和3.6~853.7nmol/L。SAR显示,两个胺基之间的碳链长度与活性息息相关,且3个和4个碳最优。靠近嘧啶端的氮原子可被氧原子取代,且对R2位而言异丙基最优,而酯基可被氰基取代(134, 代表物134a抗CQS 3D7和CQR Dd2恶性疟原虫的IC50分别为68.5和55.8nmol/L)。向苯环引入氟、氯或向间位或对位引入硝基对活性不利,但向邻位引入硝基则可提高活性。值得关注的是,绝大多数杂合体的RI<1。代表物133a(IC50:3.6nmol/L)抗CQR K1恶性疟原虫活性与青蒿琥酯(IC50:2.8nmol/L)相当,而是CQ(IC50:201.8nmol/L)的56倍。
喹啉-嘧啶杂合体135(图21, 抗CQS NF54和CQR Dd2恶性疟原虫的IC50分别为11.7~26.7nmol/L和5.2~33.3nmol/L)的抗CQS NF54和CQR Dd2恶性疟原虫活性与ART (IC50:10.0和13.0nmol/L)相当,但高于CQ(IC50:27.0和222.0nmol/L)。杂合体135的位置异构体136(抗CQS NF54和CQR Dd2恶性疟原虫的IC50分别为7.5~144.7nmol/L和4.7~74.9nmol/L)也具有良好的活性,但大多数化合物活性弱于135。对杂合体135而言,吡咯烷基、哌啶基和N-烷基哌嗪基对活性有利,而对杂合体136而言,哌啶、吗啉和硫代吗啉可提高活性。杂合体135f(IC50:12.5和5.2nmol/L)和136b (IC50:13.4和4.7nmol/L)的抗CQS NF54和CQR Dd2恶性疟原虫活性最高,优于CQ。在感染伯氏鼠疟原虫的小鼠模型中,杂合体136b(33.3mg/kg·3d, 腹腔注射)的体内活性优于135f,杂合体136b可完全治愈感染小鼠(5/5),而杂合体135f的治愈率为3/5。当给药剂量降至11.1mg/kg时,尽管杂合体136b可在小鼠无死亡的情况下完全抑制寄生虫血症,但无法完全清除小鼠体内的疟原虫。
1,2,3-三氮唑连接的喹啉-嘧啶杂合体137和138抗CQS NF54和CQR Dd2恶性疟原虫的IC50在微摩尔水平,且活性弱于CQ (IC50:18和275nmol/L)。杂合体137的活性高于138,提示喹啉和1,2,3-三氮唑之间的烷胺基对活性不利。对杂合体137而言,异丙基为R1位的最佳取代基。代表物138a(IC50:330和610nmol/L)不仅活性高,而且对VERO细胞的毒性(CC50:28.52μmol/L)低,SI为86.52。喹啉-吡唑并嘧啶杂合体139也具有潜在的抗CQS NF54和CQR K1恶性疟原虫活性,且杂合体139a,b(IC50:<3.9~7.8nmol/L)的活性可与ART(IC50:7和6.5nmol/L)媲美,而是CQ(IC50:17和347nmol/L)的>3倍。
2.11 喹啉-吡啶/吡啶酮杂合体
喹啉-吡啶杂合体140(图22)抗CQS NF54和CQR K1恶性疟原虫的IC50分别为36和33nmol/L,其抗CQS NF54恶性疟原虫活性与CQ(IC50:16nmol/L)相当,抗CQR K1恶性疟原虫活性则是CQ(IC50:194nmol/L)的5.8倍。进一步研究发现,用吡啶或苯环代替喹啉环可提高抗恶性疟原虫活性,提示喹啉环并非高活性所必需。
喹啉-羟基吡啶酮杂合体141抗CQS D10和CQR K1恶性疟原虫的SAR表明,N-1位(R1)环丙基优于甲基。与无取代衍生物相比,向R3位引入苄基对活性不利。杂合体141a(IC50:64, 47, 505和463nmol/L)和142(IC50:41, 122, 89和76nmol/L)对CQS D10, 3D7和CQR Dd2, K1恶性疟原虫的活性较高,与CQ(IC50:19, 23, 279和180nmol/L)相当。在感染伯氏鼠疟原虫的小鼠模型中,杂合体141a(每日8mg/kg, 静脉注射给药)可在给药d9降低82.9%的寄生虫血症,但弱于CQ (口服给药每日10mg/kg可降低92.0%的寄生虫血症)。
2.12 喹啉-1,3,5-三嗪杂合体
喹啉-1,3,5-三嗪杂合体143(图23)的抗CQS D6和CQR W2恶性疟原虫活性与喹啉和1,3,5-三嗪之间连接子的长度息息相关,且2个、3个和8个碳的连接子对活性有利,而4和5个碳的连接子对活性不利。活性最高的杂合体143a~c(IC50:210~480nmol/L)的抗CQS D6恶性疟原虫活性弱于ART和CQ (IC50:30和42nmol/L),但抗CQR W2恶性疟原虫活性高于CQ (IC50:420nmol/L)。进一步研究发现,用胺基烷基醇代替1,3,5-三嗪上的取代苯胺或以哌嗪为连接子(杂合体144和145)则会导致活性降低。
对苯基二胺基连接的喹啉-1,3,5-三嗪杂合体146(IC50:3.01~313nmol/L)的抗CQS 3D7恶性疟原虫活性普遍优于间苯基二胺基连接的衍生物147(IC50:26.30~291.73nmol/L),且绝大多数杂合体(IC50:<10nmol/L)的抗CQR K1恶性疟原虫活性高于CQ (IC50:802nmol/L)。有趣的是,含有氯的喹啉-1,3,5-三嗪杂合体148(IC50:118~807nmol/L)也具有良好的抗CQR W2恶性疟原虫活性。在感染CQR N-67约氏疟原虫的小鼠模型中,杂合体146a~g(腹腔注射给药25或50mg/kg, 或口服给药50或100mg/kg)可在给药d4完全抑制寄生虫血症。在杂合体146a和146b(腹腔注射给药50mg/kg或口服给药100mg/kg)组,给药d28无小鼠死亡。良好的体内外活性安全性,使得二者可作为先导物进一步优化。
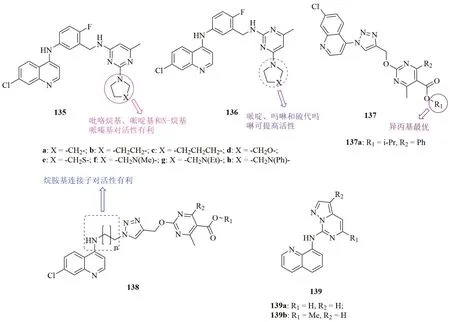
图21 喹啉-嘧啶杂合体135~139的化学结构
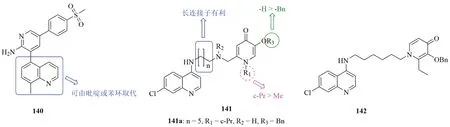
图22 喹啉-吡啶/吡啶酮杂合体140~142的化学结构
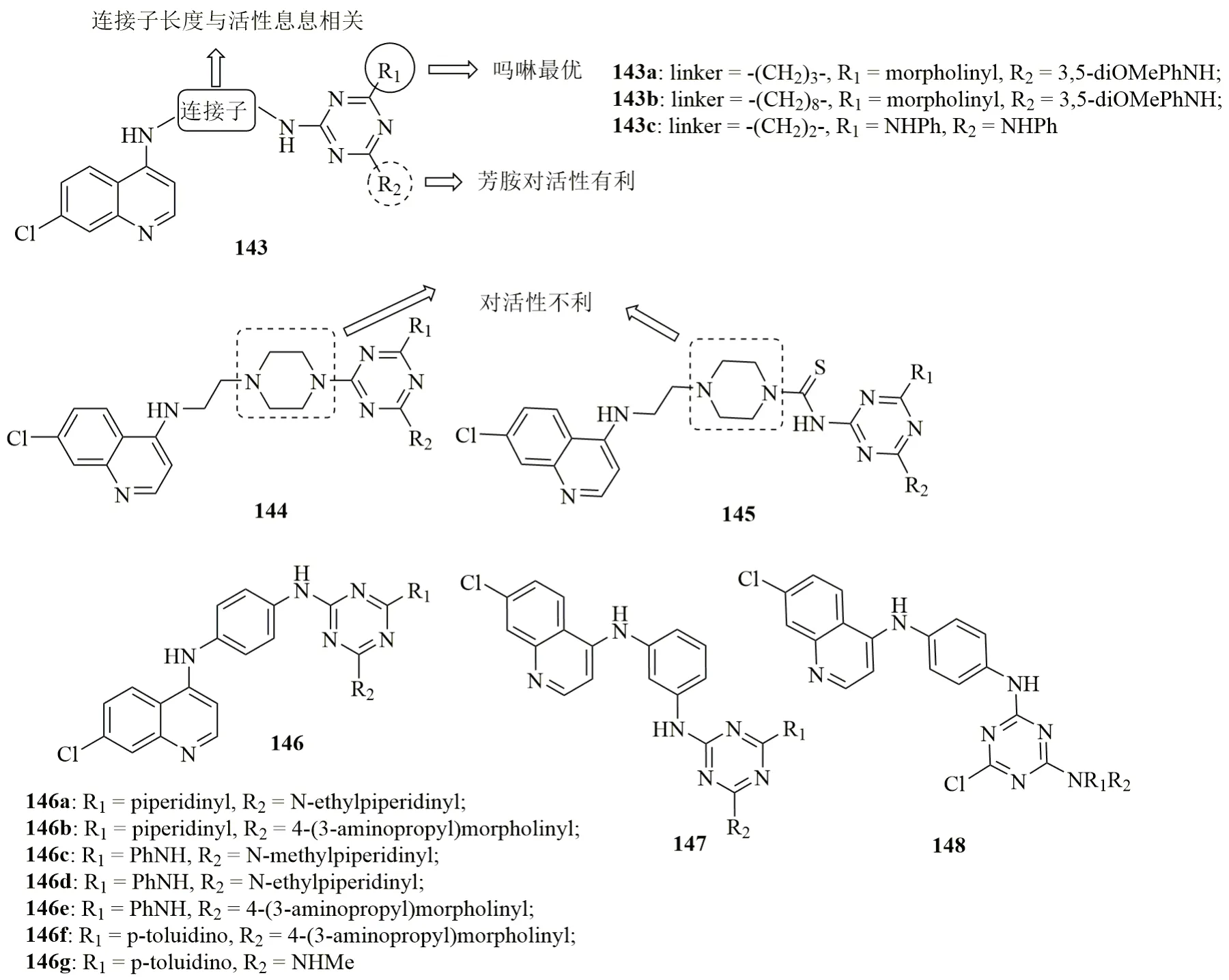
图23 喹啉-1,3,5-三嗪杂合体143~148的化学结构
2.13 其他喹啉杂合体
喹啉-磺酰胺杂合体149(图24, IC50:1160~6090nmol/L)具有中等强度的抗CQR FCR-3恶性疟原虫活性,尽管含有吸电子基如硝基的杂合体具有更高的活性,但均弱于QN(IC50:170nmol/L)。用二胺基烷基取代哌嗪可提高这类杂合体的活性,且大多数杂合体150(IC50:50~680nmol/L)具有良好的抗CQR W2恶性疟原虫活性。SAR表明,延长连接子长度对活性有利,且与无取代衍生物相比,在苯环上引入甲基和氯可提高活性。其中,杂合体150a和150b抗CQR W2恶性疟原虫的IC50分别为50和90nmol/L,活性优于CQ(IC50:460nmol/L),且对BGM细胞的毒性(CC50:~100μmol/L)极低。在感染伯氏鼠疟原虫的小鼠模型中,杂合体150a和150b(10mg/kg, 口服给药)在给药d5可抑制27%和49%的寄生虫血症,活性弱于CQ(给药20mg/kg可抑制93%的寄生虫血症)。

图24 喹啉杂合体149~154的化学结构
绝大多数喹啉-噁嗪杂合体151(抗CQS MRC-02和CQR RKL9恶性疟原虫的IC50分别为26~37.5nmol/L和43.5~130.5nmol/L)的抗CQS MRC-02和CQR RKL9恶性疟原虫活性与CQ(IC50:21.8和177nmol/L)相当或更优。杂合体151a~c(IC50:43.5~68nmol/L)的抗CQR RKL9恶性疟原虫活性最高,且在感染CQR N-67约氏疟原虫的小鼠模型中,三者(每日50mg/kg,静脉注射给药)可降低69.93%,48.52%和44.72%的寄生虫血症,但弱于CQ(完全消除)。
某些喹啉-呋喃/噻吩杂合体如152也具有潜在的抗恶性疟原虫活性,化合物152a(IC50:2,8和9nmol/L)对CQS D6,CQR W2和耐多药C235恶性疟原虫的活性极高,是CQ(IC50:15, 595和206nmol/L)的7.5~74.5倍。在感染KBG 173伯氏鼠疟原虫的小鼠模型中,杂合体152b(口服给药)在给药剂量为每日40mg/kg时会复发,但给药每日160mg/kg连续给药31d未见小鼠死亡。
喹啉-腙杂合体153的抗CQS T96和CQR K1恶性疟原虫SAR显示,羟基为苯环上的最佳取代基,且杂合体153a(IC50:249.7和45.3nmol/L)的抗CQR K1恶性疟原虫活性是CQ(IC50:89.1nmol/L)的2倍。对杂合体154而言,化合物154a和154b(IC50:21.64和22.52nmol/L)的抗CQR K1恶性疟原虫活性是CQ(IC50:495.94nmol/L)的约22倍,值得进一步研究。
3 结束语
喹啉类抗疟疾药物对疟疾的防治不可或缺,但是由于耐药疟疾的不断涌现和广泛传播,使得研发新型抗疟疾药物势在必行。杂合体分子具有克服耐药疟疾的潜力,故将喹啉与其它具有抗疟疾活性的药效团杂合是获得新型抗疟疾药物的有效途径。经过药物化学家的不懈努力,目前已发现多个对药敏型和耐药型甚至耐多药恶性疟原虫具有优秀体内外活性的喹啉杂合体。
本文将综述自2010年以来喹啉二聚体、喹啉-氨基酸、喹啉-唑、喹啉-查耳酮、喹啉-肉桂酸、喹啉-二茂铁、喹啉-吲哚/吡咯、喹啉-内酰胺、喹啉-大环内酯、喹啉-嘧啶、喹啉-吡啶/吡啶酮和喹啉-1,3,5-三嗪等喹啉类杂合体即IV型杂合体在抗耐药疟疾领域的最新研究进展,并探讨了此类杂合体的SAR,以期为进一步研究提供理论支持。
猜你喜欢
发明与创新(2020年5期)2020-12-20
科学导报(2020年69期)2020-11-09
色谱(2020年3期)2020-02-12
火炸药学报(2019年5期)2019-11-11
天然气与石油(2018年6期)2019-01-29
郑州大学学报(医学版)(2018年5期)2018-10-10
中国感染与化疗杂志(2018年6期)2018-01-19
中成药(2017年7期)2017-11-22
国外医药(抗生素分册)(2016年1期)2016-07-10
原子与分子物理学报(2014年4期)2014-02-28
