
蛋白酪氨酸磷酸酶SHP2及其抑制剂的研究进展
2019-09-23 06:39孔娇龙亚秋
药学进展 2019年7期
孔娇,龙亚秋
(苏州大学医学部药学院,江苏 苏州 215123)
蛋白酪氨酸磷酸酶(protein tyrosine phosphatase,PTP)催化磷酸酪氨酸的去磷酸化,是哺乳动物信号传导中的关键控制元件,其生物学功能的偏差会引起机体调控紊乱而导致癌症、糖尿病、自身免疫性疾病等多种疾病的发生[1]。其中,含Src同源2结构域蛋白酪氨酸磷酸酶(Src homology 2 domaincontaining protein tyrosine phosphatase,SHP2)是目前PTP家族中唯一被证实的原癌蛋白[2],是一个理想的癌症干预靶标,主要表现在3个方面:首先,SHP2是多条激活RAS信号通路的共有节点,激活RAS对癌细胞的生长和存活都非常重要,几乎所有的受体酪氨酸激酶(receptor tyrosine kinase,RTK)通过激活SHP2来启动RAS信号通路,因此一个合适的SHP2抑制剂可以对不同的RTK基因突变“一网打尽”,有潜力成为广谱抗癌药物;其次,由于蛋白酪氨酸激酶(protein tyrosine kinase,PTK)与SHP2信号通路的重叠,SHP2抑制剂可以与激酶抑制剂联用对相互连接的信号通路进行双重抑制,这种组合疗法比单一疗法更有效,既不易产生耐药性,又能逆转PTK抑制剂的获得性耐药性[3];最后,SHP2还参与程序细胞死亡检查点PD-1/PD-L1的信号通路,调节T细胞的活性[4]。鉴于当前抗PD-1/PD-L1肿瘤免疫疗法在临床上的成功应用,SHP2小分子抑制剂作为PD-1/PD-L1抗体药的重要补充,作为小分子肿瘤免疫治疗药物备受临床期待[5]。SHP2的激活突变在Noonan综合征[6]以及多种癌症类型中被发现,包括白血病、肺癌、乳腺癌以及神经母细胞瘤[7-10]。因此,SHP2是癌症治疗的潜在靶点,其抑制剂的开发已成为当前抗癌新药研究的热点。
SHP2的小分子抑制剂按照作用位点不同可分为两大类——催化位点抑制剂和变构位点抑制剂。催化位点抑制剂是靶向磷酸酶的活性位点,与酪氨酸磷酸酯底物竞争,这在高度同源性的PTP家族(如SHP1、PTP1B)中缺乏选择性,而且抑制剂必需的高电荷性官能团也导致细胞透膜性差和口服生物利用度低等问题。这也使得PTP在很长一段时间成为不可成药性靶蛋白。
最近2年,通过设计的定向高通量筛选获得的小分子变构抑制剂,通过与SHP2的非保守变构位点结合而稳定酶的非活性构象从而抑制SHP2的催化功能,因而相对其他PTP家族成员具有很好的选择性,并且具有较高活性和口服生物利用度。变构位点抑制剂的发现将SHP2抑制剂推向了临床应用。
然而,变构位点抑制剂的筛选和发现相对复杂,需要投入更多的时间和资金。本文主要综述近年来报道的SHP2抑制剂,以期为现有抑制剂的临床研发和新型SHP2抑制剂的发现提供参考。
1 SHP2的结构及其参与的信号通路
SHP2是由Ptpn11基因编码的非受体蛋白酪氨酸磷酸酶,含有2个串联的Src同源结构域(N-SH2/C-SH2)、催化位点结构域(PTP)和带有2个磷酸化酪氨酸位点的C-末端尾部。1998年,Barford及其同事报道了完整的SHP2蛋白晶体结构[11]。晶体结构显示,在非活性状态下,SHP2蛋白的N-SH2结构域与PTP结构域结合并阻断其底物进入催化位点,从而导致SHP2活性受到抑制。当SH2结构域与特定的磷酸酪氨酸基序(motif)结合时,SHP2的自抑制状态被破坏,处于活化开放构象,发挥将底物去磷酸化的功能(见图1)。
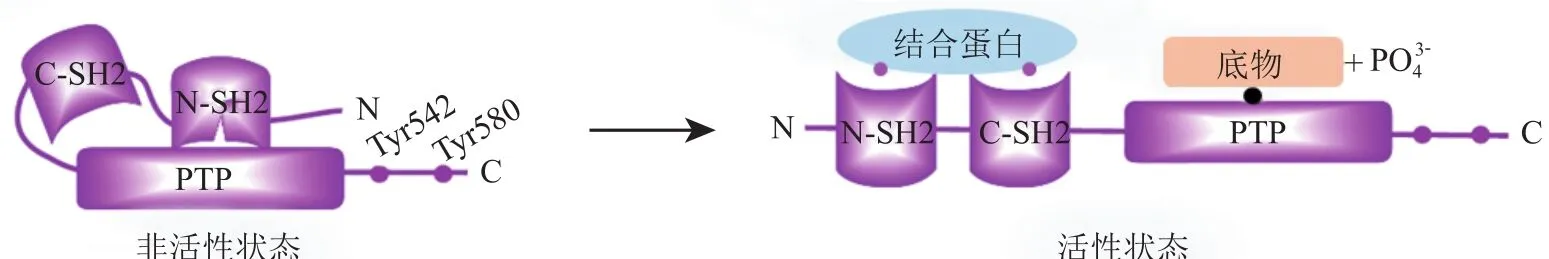
图 1 SHP2蛋白的激活[2]Figure 1 Activation of SHP2
SHP2参与调节生物体内多种信号通路。SHP2结合位点存在于RTK和骨架衔接子(如GAB、IRS、FRS等蛋白)中,因此这种“分子开关”确保SHP2仅在适当的细胞区域被激活[12]。在生长因子和细胞因子信号传导中,SHP2作用于RAS的上游,并使细胞外调节蛋白激酶(extracellular regulated protein kinases,ERK)/丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)途径完全活化[13-14]。此外,SHP2的C末端酪氨酸能够响应大多数激动剂而发生磷酸化,酪氨酸磷酸化的SHP2募集接头蛋白GRB2和鸟嘌呤核苷酸交换因子SOS,有助于RAS活化。此外,SHP2通过其N-SH2结构域结合免疫检查点蛋白PD-1的磷酸酪氨酸基序(pTyr motif),参与调节T细胞的活性(见图2)。能够阻断PD-1与SHP2之间蛋白-蛋白相互作用的抑制剂有望成为小分子肿瘤免疫疗法,因此SHP2也成为肿瘤免疫治疗中一个有潜力的药物靶点[15-17]。
2 SHP2小分子抑制剂
2.1 催化位点抑制剂
SHP2的作用底物为酪氨酸磷酸酯,因此其催化位点抑制剂大多含有模拟磷酸酪氨酸的离子官能团。这些离子官能团作为磷酸酪氨酸模拟物可结合到SHP2的底物催化口袋,从而起到对SHP2活性的抑制作用。SHP2催化位点抑制剂根据其含有的离子官能团不同,可分为含磺酸基团和含水杨酸基团的抑制剂;另外,天然产物中也发现了活性位点导向的SHP2抑制剂。
2.1.1 含磺酸基团的抑制剂 化合物NSC-87877(1)是第1个被成功鉴定出能够抑制SHP2 PTP结构域的抑制剂 (IC50= 0.32 μmol·L-1),由 Chen 等[18]通过对美国国家癌症研究所建立的多元化的化合物库筛选得到。分子模拟和定点突变研究表明NSC-87877与SHP2 PTP的催化裂缝结合。其对SHP2的选择性高于其他PTP家族成员(PTP1B、HePTP、DEP1、CD45和LAR),但对同源性酶SHP1显示出几乎同等的抑制活性。细胞测试表明:NSC-87877抑制表皮生长因子(epidermal growth factor,EGF)诱导的RAS和ERK1/2的活化,但不阻断EGF诱导的GRB2结合蛋白1 (GRB2 associated binding protein 1,GAB1)酪氨酸磷酸化或GAB1-SHP2结合。他们的研究首次提供SHP2介导的EGF诱导ERK 1/2、MAPK活化的药理证据。
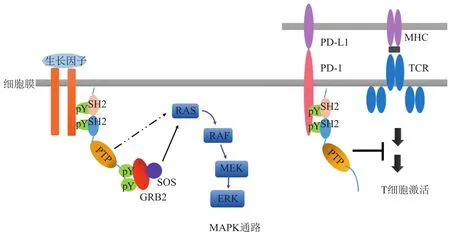
图2 SHP2 介导的信号通路[12]Figure 2 SHP2-mediated signaling pathways
Hellmuth等[19]通过高通量虚拟筛选得到化合物PHPS1(2),系第1个SHP2特异性抑制剂(Ki= 0.7 μmol·L-1),其对PTP1B和SHP1分别显示8倍和15倍的选择性。他们使用重组SHP2的底物滴定研究表明PHPS1是SHP2的竞争性抑制剂。值得注意的是,PHPS1抑制细胞中由SHP2依赖性激活的RAS/MAPK途径,并且PHPS1不影响PMA(Phorbol-12-myristate-13-acetate)或致癌RAS诱导的非依赖性SHP2的ERK1/2活化。PHPS1还有效阻断人肿瘤细胞的增殖和不依赖锚定的生长(anchorage-independent growth),并且对正常上皮细胞无细胞毒性。
张仲寅课题组[20]从现有且具有良好药动学特征和可耐受副作用的化合物库中筛选出第3代β-内酰胺类抗生素头孢磺啶(3),作为可逆的竞争性SHP2 抑制剂 [IC50=(16.8 ± 2.0)μmol·L-1],其对SHP1和PTP1B显示2倍和11倍的选择性。X射线晶体学研究和结构分析确定头孢磺啶中的磺苯基乙酰胺为磷酸酪氨酸模拟物,在此基础上,该课题组采用结构导向和基于片段的方法发现了几种含有磺苯基乙酰胺官能团的SHP2抑制剂,IC50均在低微摩尔范围内,且对SHP1的选择性提高了5倍。其中化合物4可以特异性地抑制SHP2介导的信号传导,并且抑制MDA-MB-231和ErbB2阳性的乳腺癌SKBR3细胞以及肺癌H1975细胞的生长。
2.1.2 含水杨酸基团的抑制剂 张仲寅课题组[21]利用计算机虚拟筛选发现天然产物水杨酸可以作为磷酸酪氨酸模拟物。他们运用点击化学(click chemistry)的方法构建了一类含吲哚水杨酸结构单元的化合物库,经过一系列构效关系研究,最终发现化合物5是有效的SHP2可逆非竞争性抑制剂。其具有较好的细胞活性[IC50=(5.5 ± 0.4)μmol·L-1],能够阻断HEK293细胞中由生长因子刺激的ERK1/2激活,抑制由粒细胞巨噬细胞集落刺激因子(granulocyte macrophage colony stimulating factor,GM-CSF)刺激的SHP2功能获得性突变诱导的造血细胞过度增殖。化合物5与催化结构域SHP2的X射线晶体结构分析显示水杨酸基团占据PTP活性位点,联苯基团与PTP之间非保守区域发生疏水相互作用(见图3),但该化合物对SHP1和PTP1B仅有3倍的选择性。
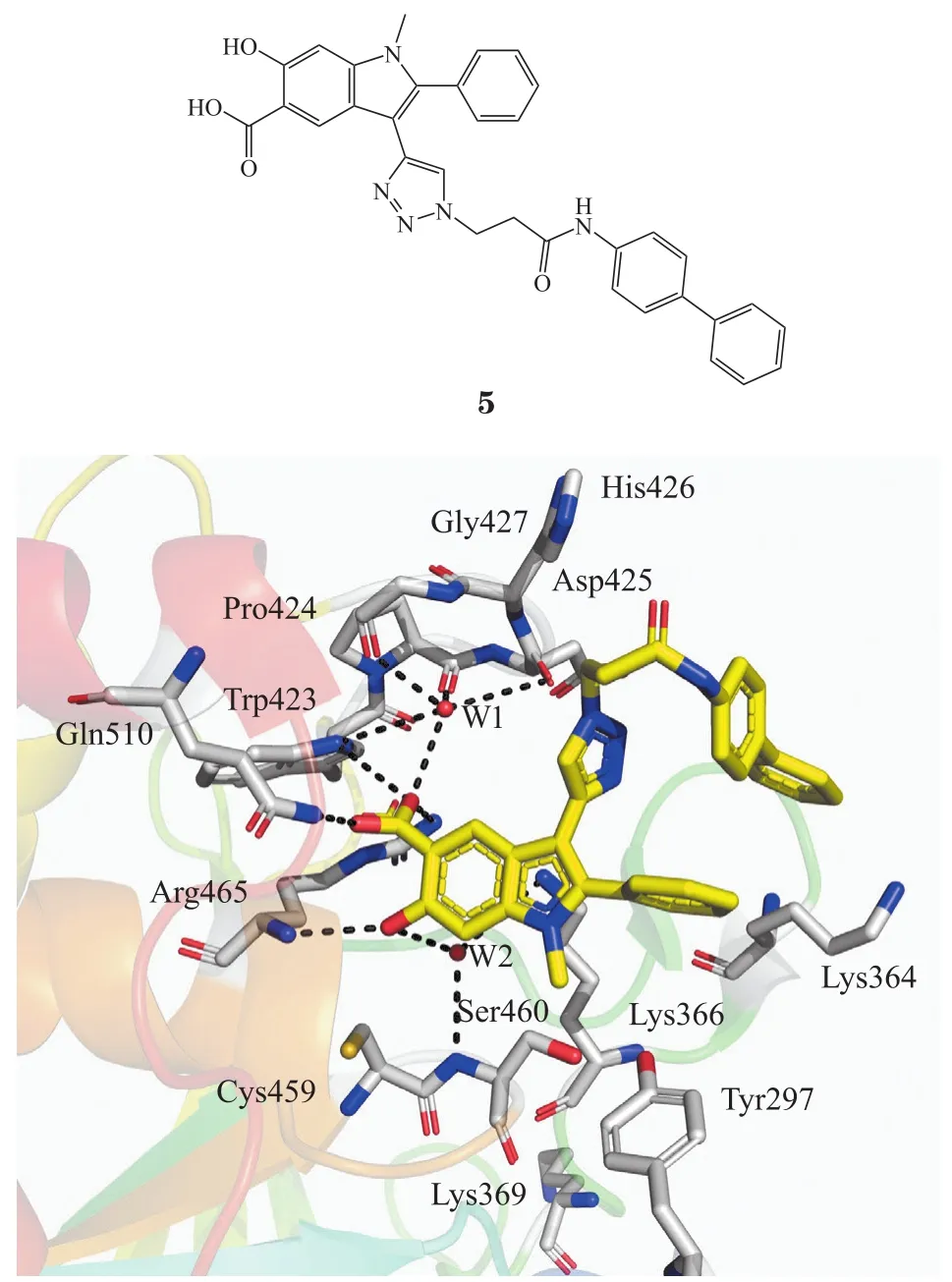
图3 化合物5与SHP2蛋白晶体结构[21]Figure 3 Crystal structure of compound 5 and SHP2 protein
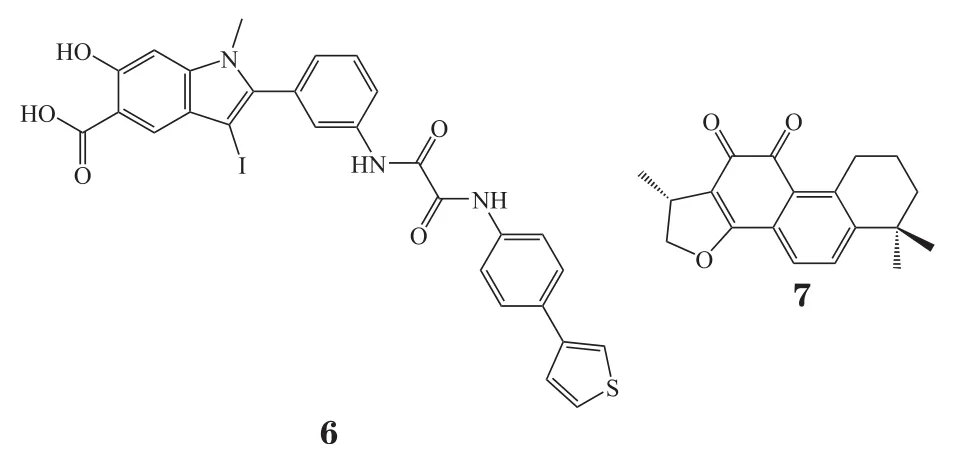
为了发现具有选择性的SHP2抑制剂,张仲寅课题组[22]采用结构导向的方法,以羟基吲哚羧酸作为SHP2活性位点的靶点,并依据基于片段的药物发现策略,引入额外的官能团,旨在与靠近催化口袋的周边位点相互作用,以提高SHP2的结合亲和力和选择性。在化合物5的基础上,经过一系列结构优化,他们发现化合物6对SHP2的选择性是其同源物SHP1和PTP1B的7倍和11倍,结构分析和分子模拟揭示羟基吲哚羧酸结合在SHP2活性位点,而草酰胺连接子和苯基噻吩基团与β5-β6环中的残基发生相互作用,这使得化合物6对SHP2有较好的活性[IC50=(0.2 ± 0.02)μmol·L-1]和选择性。细胞实验中,化合物6可特异性地阻断细胞内的SHP2依赖性信号传导。此外,其还减弱生长因子介导的ERK1/2和蛋白激酶B(protein kinase B,PKB或AKT)活化,并在非小细胞肺癌细胞系H1975、ErbB2阳性的乳腺癌细胞系SKBR3和致癌基因KITD814V表达的32D骨髓细胞中表现出优异的抗增殖活性。
2.1.3 源于天然产物的抑制剂 Liu等[23]通过计算机虚拟筛选与实验测定,从天然产物数据库中筛选出的隐丹参酮(7)可结合SHP2催化位点结构域(IC50=22.5 μmol·L-1),对 SHP1 选择性约为 1.76 倍。而且,化合物7对SHP2 E76K突变蛋白也有相同程度的抑制(IC50=23.90 μmol·L-1),而对野生型全长的 SHP2 蛋白活性较弱(IC50=45.18 μmol·L-1)。酶促动力学分析表明,化合物7是一种混合型和不可逆的SHP2抑制剂。在细胞实验中,化合物7通过抑制SHP2催化活性而使RAS-EAK、PI3K-AKT和JAK2-STAT5信号通路下调。此外,SHP2中具有活化突变E76K的小鼠骨髓祖细胞和患者白血病细胞对该抑制剂敏感。不过目前尚未培养出该抑制剂与SHP2蛋白的晶体结构。
2.2 变构位点抑制剂
SHP2催化位点抑制剂经过十几年的发展,仍然没有达到理想的体内疗效。虽然活性最好的催化位点抑制剂化合物6在黑色素瘤模型中显示出良好的前景[24],但是最近的研究已经质疑这种催化位点抑制剂在细胞环境中的特异性[25]。于是,科学家们将研究重点转向SHP2的变构模式,由此开启了SHP2抑制剂研究的新阶段。2016年,诺华研究团队首次报道了作用于SHP2的3个结构域界面的变构位点抑制剂,截至2019年4月份,已有10余种结构类型的有效SHP2变构位点抑制剂被报道,其中多数为诺华公司的研究成果。以下将按研究团队分类对SHP2变构位点抑制剂进行介绍。
2.2.1 诺华团队报道的抑制剂 SHP099(8)是由诺华公司报道的首个作用于SHP2变构位点的抑制剂[26-27]。该团队设计了新颖的药物筛选方法:以 DIFMUP(6,8-difluoro-4-methylumbelliferyl phosphate)为底物[28],双磷酸化酪氨酸IRS-1肽激活SHP2,从10万个化合物中筛选出可以抑制全长SHP2蛋白(氨基酸残基1-525)的抑制剂,从中去除作用在PTP区域(氨基酸残基237-529)的抑制剂,得到化合物SHP836(9),其对于全长SHP2蛋白的IC50为12 μmol·L-1,并且针对PTP结构域的IC50大于 100 μmol·L-1。通过 X 射线晶体学发现,SHP836不与PTP催化位点结合,而是与C-SH2、N-SH2和PTP结构域之间形成的隧道样口袋结合,稳定了SHP2的自抑制构象。采用基于结构的药物设计对SHP836进行优化和变形,最终发现了以氨基吡嗪为母核的化合物SHP099是一种高效、可口服的小分子 SHP2 抑制剂(IC50= 0.071 μmol·L-1),但对其他PTP家族(包括SHP1)和激酶没有显著活性。根据 SHP099与SHP2蛋白的晶体复合物,SHP099同时结合N-SH2、C-SH2和PTP结构域的界面,与3个结构域中的关键氨基酸残基(Arg111、Phe113和Glu250)产生氢键相互作用,从而通过变构机制抑制SHP2活性(见图4)。相应地,变构口袋的双突变体(SHP2T253M/Q257L)会破坏SHP099与SHP2蛋白的结合。SHP099在体外抑制RAS/ERK信号传导以抑制RTK驱动的人癌细胞的增殖。动物实验中,通过经口给药方式,SHP099在小鼠异种移植模型中表现出剂量依赖性抗肿瘤活性,且无明显毒副作用。
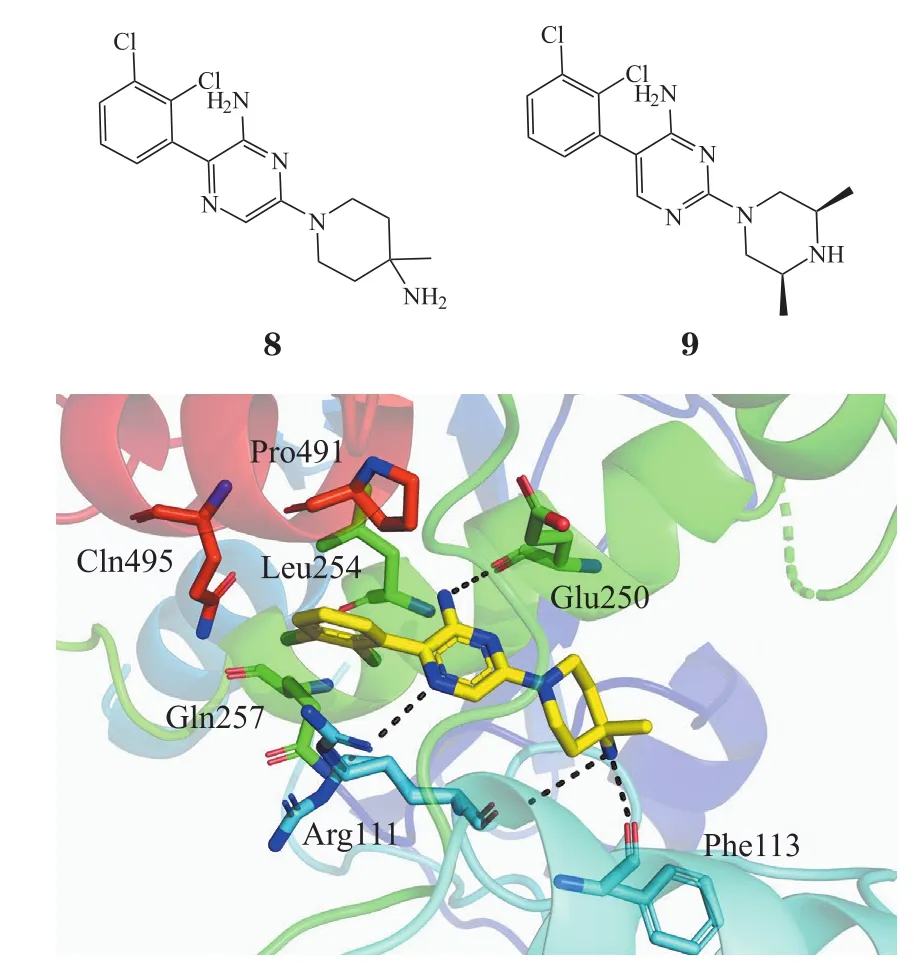
图4 SHP099作用于SHP2蛋白的关键残基[26]Figure 4 Key residues involved in the interaction of SHP099 with SHP2 protein
随后,诺华研究团队使用Maestro中的SiteMap分析SHP2蛋白中可配位的口袋,并揭示了3个潜在的小分子变构结合位点(见图5)[29],分别为:1)先前报道的SHP099的变构结合位点,即“隧道”结合位点;2)离“隧道”约20 Å(1 Å=10-10m)的“门闩”结合位点;3)蛋白质另一面的“凹槽”结合位点。考虑到多个变构口袋存在的可能,他们使用突变型SHP2蛋白(SHP2T253M/Q257L)构建了新的抑制剂筛选模式,同样以DIFMUP为底物,双磷酸化酪氨酸IRS-1肽激活SHP2,筛选了约150万个化合物。用近全长的SHP2蛋白(SHP21-525)鉴定具有抑制模式的化合物,再利用SHP2PTP区域去除活性位点抑制剂。为了除去与SHP099作用在同一变构位点的抑制剂,他们选择了对SHP2T253M/Q257L仍具有抑制活性的化合物,并使用X射线晶体学来确定结合模式以及采用差示扫描荧光法(differential scanning fluorimetry,DSF)来研究热稳定性,最终发现了一系列具有未知抑制模式的化合物,其中包括以三唑并喹唑啉酮为母核的化合物SHP244(10)。SHP244对SHP2有微弱抑制活性(SHP21-525:IC50=60 μmol·L-1,SHP2PTP:IC50> 100 μmol·L-1),X 射线晶体学显示 SHP244 与之前预测的位于N-SH2和PTP结构域之间的“门闩”结合位点结合。他们基于结构的衍生化设计合成了化合物 SHP844(11,SHP21-525:IC50=18.9 μmol·L-1,SHP2PTP:IC50> 100 μmol·L-1)和化合物 SHP504(12,SHP21-525:IC50= 21 μmol·L-1,SHP2PTP:IC50> 100 μmol·L-1)用于对SHP2的生物学评价,发现这2个化合物在KYSE-520癌细胞中观察到MAPK信号通路下游标记物DUSP6 mRNA的下调。
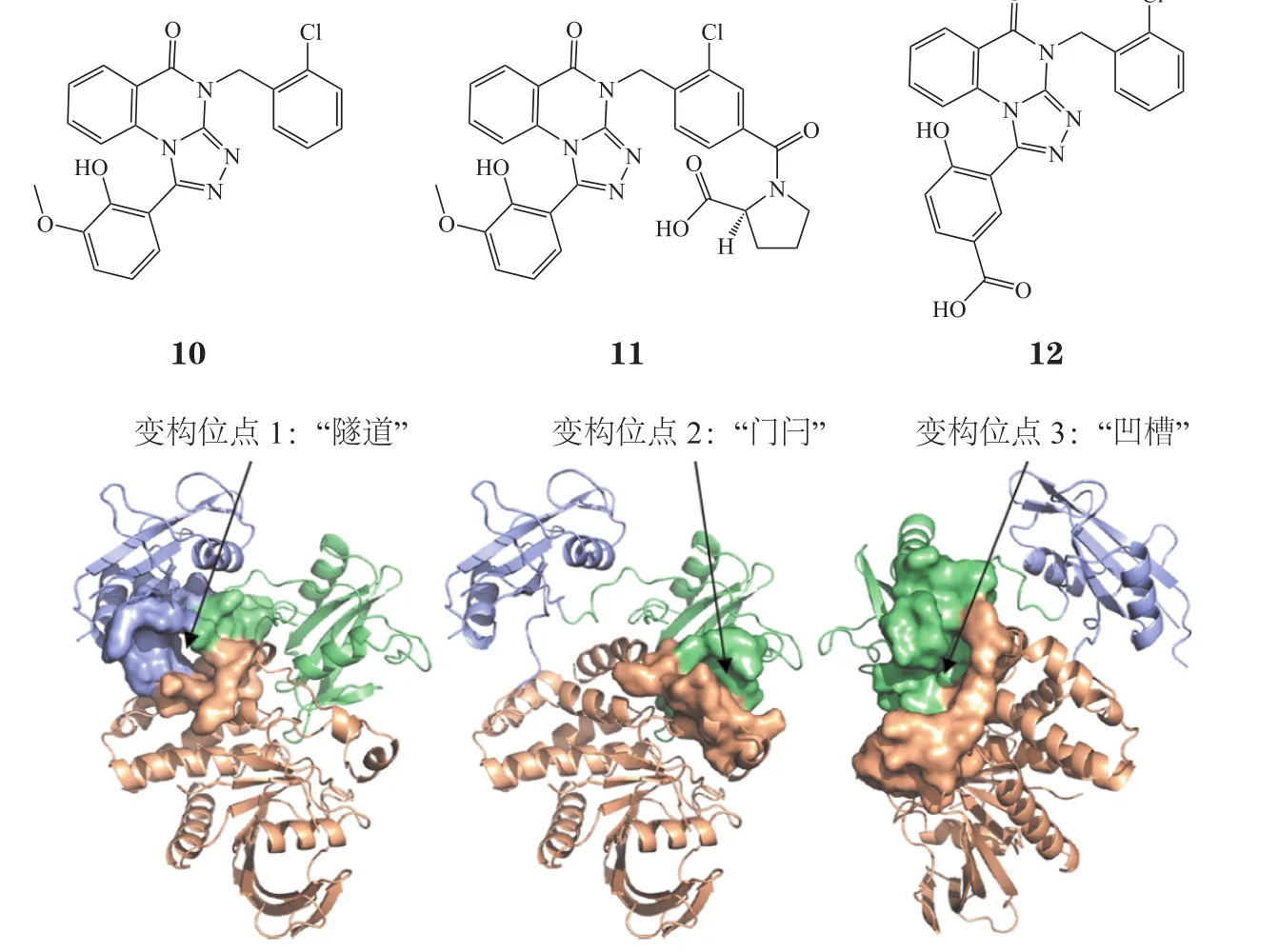
图5 SHP2自抑制构象的变构口袋预测[29]Figure 5 Allosteric pocket prediction of SHP2 self-suppressed conformation
最近,诺华研究团队连发2篇文章报道了新型结构的SHP2变构抑制剂[30-31]。他们将SHP099结构中的药效基团与经过高通量筛选得到的化合物相融合,成功发现了以吡唑并嘧啶酮为母核的化合物13,X射线晶体学显示该化合物与SHP099一样,均结合在SHP2的“隧道”位点。通过对化合物13进行胺取代基的延伸和构象限制得到化合物14,与化合物13相比,化合物14显著增强了由SHP2介导的ERK激酶磷酸化的抑制作用及在KYSE520细胞系的抗增殖作用。为了改善化合物对hERG的选择性,他们又对侧链氯代芳烃进行优化,得到化合物SHP389(15),既保留了较好的生物活性,又改善了hERG的选择性,但其口服生物利用度低(约2%),无法在小鼠肿瘤异种移植模型中作进一步的药理学评价。为了获得更理想的SHP2变构抑制剂,诺华研究团队结合化合物13、16以及SHP099的结构特点进一步进行构效关系研究,他们将融合的双环系统变成单环嘧啶酮母核,最终发现SHP394(17)是有效的SHP2抑制剂。当对皮下植入Detroit-562肿瘤细胞的免疫受损小鼠经口给予SHP394时,其显示出剂量依赖性抗肿瘤能力。通过对芳基硫醚和螺环胺部分构效关系研究合成了一系列具有与SHP394相似的抗增殖效力的化合物。其中,化合物18对SHP2的活性有所改善,化合物19则改善了口服吸收和hERG选择性。
2.2.2 其他研究团队报道的抑制剂 Revolution Medicines公司一直致力于开发靶向SHP2的变构抑制剂,其中,RMC-4630就是该公司研发的口服有效的 SHP2小分子变构抑制剂,目前已进入临床试验阶段。RMC-4630的结构尚未公布,但其同系化合物RMC-4550(20,IC50= 0.583 nmol·L-1)的药效和作用机制被充分研究并报道。SHP2抑制剂RMC-4550对具有RAS-GTP依赖的致癌性BRAF(如第3类BRAF突变体)、NF1缺失或核苷酸循环致癌RAS(如KRASG12C)的人类癌症模型有效[14]。RMC-4550通过破坏SOS1介导的RAS-GTP来降低致癌RAS/RAF/MEK/ERK信号传导和癌症生长。研究结果阐明了SHP2在RAS-GTP依赖性致癌BRAF、NF1缺失和核苷酸循环致癌KRAS的癌症中促进RAS/MAPK途径激活的关键功能。对于携带这些致癌驱动因子的癌症患者,抑制SHP2是一种有前景的分子治疗策略。
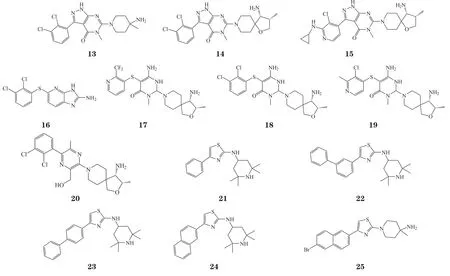
目前,针对野生型SHP2蛋白的变构抑制剂已经取得很大进展,但对于一些致癌性SHP2突变蛋白(如SHP2E76K、SHP2G60V、SHP2S502P等),变构抑制剂效果并不理想[32]。Xie等[33]开发了靶向SHP2E76A蛋白的变构抑制剂。他们从约2万个化合物中筛选出针对SHP2E76A蛋白有抑制作用的化合物,再从中除去作用在SHP2PTP区域的化合物,发现化合物21对SHP2E76A的 IC50为 19.1 μmol·L-1,但对 SHP2PTP没有影响。他们推测脂肪胺基团可能存在氢键相互作用,通过用疏水性增强的联苯或萘基团替代苯环,同时保持脂肪胺基团,得到活性提高的抑制剂 22~24,IC50分别为 3.27、5.3 和 2.55 μmol·L-1。化合物 22 与SHP2E76A蛋白的晶体复合物揭示与化合物SHP099一样,该化合物与SHP2E76A结合在相同的变构口袋,但存在不同的作用方式。接着,他们对化合物22进行构效关系研究以提高生物活性,发现将哌啶环上的氨基裸露出来,以及在萘环中引入吸电子取代基可提高化合物活性,最终得到化合物25,其对SHP2E76A的 IC50为 0.7 μmol·L-1,可有效抑制癌细胞中的ERK1/2和AKT活化。重要的是,化合物25抑制Yes相关蛋白(Yes-associated protein,YAP)依赖的荧光素酶报告基因和YAP癌基因的表达,表明YAP活性受SHP2PTP催化功能调控。化合物25抑制多种癌细胞系的增殖并抑制肺癌细胞H1975的集落形成,其在MV4-11异种移植模型中表现出良好的药动学特性和抗肿瘤活性。
Wu等[34]利用计算机辅助药物设计数据库筛选与细胞检测相结合,发现了新型SHP2抑制剂(26),计算机模拟显示化合物26的结构很好地补充了对接模拟中SHP2 C-SH2与PTP结构域之间的结合口袋(见图6),分子动力学模拟表明SHP2蛋白的运动在与化合物26结合时受到抑制,进一步支持其稳定了SHP2的自身抑制构象。值得注意的是,化合物26对携带活化突变SHP2E76K的小鼠和人白血病细胞抑制效果显著,但对SHP2E76K和SHP2WT蛋白的活性无显著差异。遗憾的是,研究者没有解析出该化合物与SHP2蛋白的晶体结构。
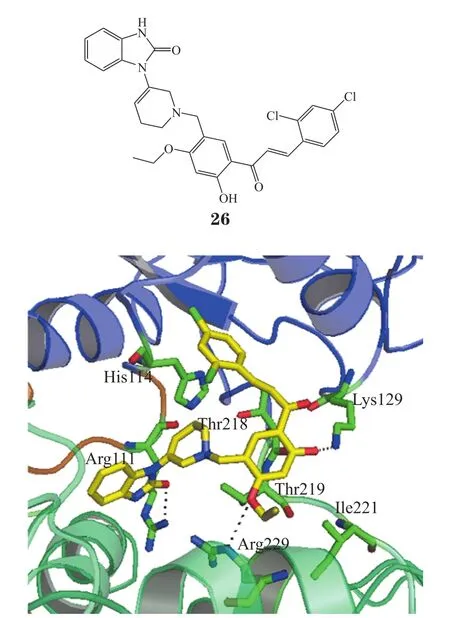
图 6 化合物 26 与SHP2的模拟对接图[34]Figure 6 Binding mode of compound 26 with SHP2 by autodocking
Brennan 等[35]认为尽管稳定SHP2自身抑制的小分子化合物(如SHP099)对于靶向涉及SHP2的癌症具有巨大前景,但是这些化合物对于癌症相关的SHP2活化突变体的抑制活性大为减弱,因为SHP2蛋白突变本身会破坏其自身抑制构象。但是,直接作用在SHP2的PTP结构域上的变构位点抑制PTP活性的化合物将提供变构SHP2靶向的替代模式。该课题组先前已经证明SHP2催化结构域含有一个隐藏的变构抑制位点,并且该位点在大多数其他PTP家族成员中不存在。这一发现源于意外观察到SHP2被双砷化合物 FlAsH(27)所抑制,进一步研究显示SHP2中隐藏的变构位点对FlAsH敏感性取决于位于活性位点之外的非保守半胱氨酸残基(Cys333)的存在。但 FlAsH会非特异性地与大量富含半胱氨酸的蛋白质结合,不能作为开发SHP2催化结构域变构抑制剂的先导化合物。因此,该课题组转而开发靶向Cys333的特异性共价抑制剂。他们合成了先前报道过的、可以与蛋白激酶共价结合的带有氰基丙烯酰胺基团的化合物库,从中筛选可以抑制野生型SHP2 PTP结构域,而对突变型(Cys333Pro)SHP2 PTP结构域无抑制活性的化合物,以期能够找到特异性作用在该变构口袋的抑制剂。通过筛选,他们发现化合物28对野生型SHP2的抑制活性明显高于SHP2C333P,简单的构效关系研究发现化合物28的喹啉环是必需的,将氰基丙烯酰胺替换成丙烯酰胺得到化合物29,其对野生型 SHP2 PTP 结构域的 IC50为 35 μmol·L-1,并且化合物29保留了化合物28对SHP2C333P的特异性,即使在测定的最高化合物浓度下(150 μmol·L-1),对SHP2C333P都没有显著抑制作用。LC-MS/MS分析显示,20%的化合物29会共价不可逆地作用于Cys333,不会与SHP2中保守的半胱氨酸残基(Cys459、Cys367)结合。除Cys333之外,化合物29还作用在蛋白表面或环上的其他非保守半胱氨酸残基(Cys259、Cys318和Cys486),但这些作用可能对SHP2活性没有显著影响。总之,化合物29与SHP2中非保守半胱氨酸残基Cys333作用的能力证明了用共价抑制剂靶向SHP2变构位点的原理,为开发高效、特异性的SHP2变构位点共价抑制剂提供了基础。
3 总结与展望
SHP2作为致癌酪氨酸磷酸酶受到研究者的广泛关注。最初,研究者关注靶向SHP2 PTP催化区域的抑制剂,但由于PTP区域高度保守,以及极性和带正电的环境,导致活性位点抑制剂缺乏选择性,且细胞透膜性和口服生物利用度差,所以SHP2抑制剂一直没能推进到临床。自2016年由诺华公司报道第1个变构位点抑制剂以来,已有至少2个小分子抑制剂进入临床研究,包括由诺华公司开发的TNO155和由Revolution Medicines/Sanofi 开发的RMC-4630,目前都处于Ⅰ期临床阶段,用于治疗实体瘤,但结构尚未披露。
由于SHP2的激活突变在多种疾病中被发现,但尚未开发出更有效的抑制SHP2活化突变体的抑制剂。靶向不同变构位点的抑制剂的组合可能是抑制SHP2致癌突变体的一种策略。另外,由于蛋白激酶与SHP2信号通路的重叠,SHP2抑制剂可以与激酶(MEK和ALK)抑制剂联用对相互连接的信号通路进行双重抑制[4,36],这种组合疗法比单一疗法更有效,并且不易产生耐药性。因此,开发出高效、高选择性的SHP2抑制剂以及SHP2抑制剂与激酶抑制剂联用将成为SHP2抑制剂研究的主要方向。
猜你喜欢
湖北农业科学(2022年11期)2022-07-18
电气技术(2022年5期)2022-05-23
保健与生活(2022年5期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
汽车工程师(2021年12期)2022-01-18
皮肤病与性病(2021年3期)2021-07-30
实用肿瘤学杂志(2020年4期)2020-12-08
第一财经(2019年8期)2019-08-26
作文·初中版(2017年6期)2017-06-16
恋爱婚姻家庭·青春(2016年10期)2016-10-10
