
盐酸克林霉素胶囊在中国健康受试者的药动学及生物等效性研究
2019-09-24 09:18王晶晶郁继诚武晓捷曹国英何金杰施耀国程洁如丁玲玲刘晓雪龚玉秀
中国感染与化疗杂志 2019年5期
王晶晶, 郁继诚, 武晓捷, 曹国英, 梁 虹, 金 逸, 何金杰, 张 菁, 施耀国,程洁如, 丁玲玲, 刘晓雪, 龚玉秀
克林霉素早在1966 年由Magerlein等以氯原子取代林可霉素分子中的第7位羟基首次合成而得,其作用于敏感菌核糖体的50S亚基,阻止肽链的延长,从而抑制细菌细胞的蛋白质合成[1]。克林霉素主要用于治疗敏感革兰阳性球菌和厌氧菌引起的感染,是治疗金黄色葡萄球菌骨髓炎的首选药物。其对需氧革兰阳性球菌有较高抗菌活性,如葡萄球菌属(包括耐青霉素及甲氧西林敏感株)、溶血链球菌、草绿色链球菌、肺炎链球菌等;对厌氧菌亦有良好的抗菌作用,如杆菌属、梭形杆菌属、放线菌属、消化球菌、消化链球菌等[2]。口服盐酸克林霉素胃肠道吸收迅速,达峰时间为0.75~2 h,蛋白结合率高达85%~94%,广泛分布于除脑脊液外的体液和组织中,在骨组织、胆汁、尿液、胰液中可达高浓度[3-4],也能透过胎盘,进入胎儿循环。该药在肝脏代谢,部分代谢物具抗菌活性。约给药量的10%以活性成分形式由尿排出,3.6%以活性成分形式由粪便排出。血浆消除半衰期(t1/2β)成人为2.4~3.0 h,儿童为2.5~3.4 h。肾衰竭及严重肝脏损害者t1/2β略有延长(3~5 h)。多次给药未见药物蓄积现象。血透及腹透不能清除克林霉素[5-6]。原研产品美国法玛西亚普强公司(Pharmacia & Upjohn Co)生产的盐酸克林霉素胶囊(CLEOCIN HCL)于1975年在美国上市。20 世纪80 年代中期克林霉素进入中国市场,已上市多年。国产盐酸克林霉素由上海新亚药业闵行有限公司研发。本研究通过比较中国健康受试者空腹和餐后口服国产盐酸克林胶囊(受试制剂)与原研产品CLEOCIN HCL(参比制剂),评价受试制剂与参比制剂的生物等效性。
1 材料与方法
1.1 药品与仪器
受试制剂盐酸克林霉素胶囊:规格300 mg,10粒/板,2板/盒,批号170901,含量101.8%,有效期2019年9月,上海新亚药业闵行有限公司生产。参比制剂盐酸克林霉素胶囊:规格300 mg,100粒/瓶,批号SGYP,含量101.2%,有效期2019年11月,Pharmacia&Upjohn Co.生产。克林霉素标准品:批号130422-201306,纯度84.9%,中国食品药品检定研究院。内标克林霉素-d3盐酸盐(Clindamycin-d3 hydrochloride):批号9-AKS-149-2,纯度96%,失效期2020-01-25,Toronto Research Chemicals提供。
AB Sciex API 4000 QTRAP质谱仪为美国应用生物系统公司产品;UFLC 20-AD超快速液相系统为日本岛津公司产品。
1.2 研究设计
本研究采用单中心、随机、开放、单剂量、两周期、两交叉、空腹或餐后给药试验设计。由筛选期(-28~-1 d)及试验期组成,其中试验期分为第一周期(2 d)、清洗期(7 d)和第二周期(2 d)。筛选期选择合格的健康成年受试者24名,每种性别不少于总人数的1/3,随机分配至A(受试制剂/参比制剂)、B(参比制剂/受试制剂)两个序列组中,按顺序交叉给药。本研究由复旦大学附属华山医院国家药物临床试验机构承担,研究方案经复旦大学附属华山医院伦理委员会审批通过。
1.3 受试者
所有受试者均在签署书面知情同意书后进入试验。经询问病史,无心、肝、肾、内分泌、消化道、免疫系统、呼吸系统、代谢异常、肿瘤和重症肌无力等病史,无克林霉素或林可霉素、青霉素、阿司匹林药物过敏史或过敏体质,无烟、酒嗜好,无药物滥用史,3个月内无激素替代治疗,28 d内无任何可能与克林霉素有相互作用药物应用史,3个月内无>400 mL出血或献血史,非妊娠或哺乳期妇女,无48 h内服用影响肝脏代谢酶的食物或饮料者;经实验室检查包括血常规、尿常规、肝功能、肾功能、血脂、血液妊娠检查,体格检查,静息状态生命体征和12-导联心电图检查均正常或无临床意义异常,乙醇呼吸检查和烟检(尿可替宁检测)阴性。试验期间统一饮食,禁止吸烟、饮酒、摄入饮料,禁止剧烈运动。
1.4 给药方法
受试者于每个周期给药前1 d入住病房。服药前禁食至少10 h,空腹组受试者空腹单次口服受试制剂或参比制剂,240 mL温水送服。餐后组受试者于服药当天早上07:30左右开始进食高脂高热量早餐,要求须在30 min内进餐完毕。开始进餐后30 min准时单次口服受试制剂或参比制剂,240 mL温水送服。服药前1 h及服药后1 h内禁止饮水,服药后4 h内禁食,服药后4、10 h统一进食标准午餐和晚餐。第一周期受试者服用受试制剂或参比制剂,则第二周服用参比制剂或受试制剂,过程与第一周期相同。
1.5 血样采集及处理
每个周期于给药前0 h(服药前3 h内,视为0 h)和服药后0.25、0.5、0.75、1、1.25、1.5、2、2.5、3、4、5、6、8、12、16、24 h采集受试者前臂静脉血4 mL置于EDTA-K2抗凝剂真空负压采血管中,轻柔颠倒混匀5~6次,采集后即刻置于冰浴中,于1 h内在2~8 ℃、1 500~2 000 g离心10 min,分离出血浆分装于2支冻存管中,1 mL分装于检测管中,剩余量分装于备份管中,2 h内放置超低温冰箱[(-70±10)℃]冻存,直至样本转运。转运过程全程冷链监控(约-80 ℃)。
1.6 测定方法
1.6.1 测定条件 色谱条件:色谱柱为ACE 3 C18-AR (50 mm×2.1 mm);流动相A为0.4%甲酸水溶液(pH 3.2);流动相B为乙腈;梯度洗脱;柱温为40 ℃;流速为0.4mL/min;自动进样器温度为4 ℃,进样量2 μL。质谱条件采用正离子电喷雾离子化(ESI)电离模式,多反应监测模式(MRM)扫描模式。克林霉素离子对m/ z 425.3→m/z 126.2,内标克林霉素-d3离子对m/ z 428.3→m/z 129.2,碰撞能量为40 eV。
1.6.2 样本前处理 血浆样本室温完全融化后混匀30 s,取50 μL样本置于96孔板中,加入20 μL 内标工作溶液(内标 500 ng/mL,甲醇∶水=1∶1)涡旋混匀,加入500 μL甲醇溶液,涡旋3 min,4 ℃、3 200 g离心5 min,取上清液50 μL至新的96孔板中,40 ℃氮气吹干,加入250 μL复溶液[乙腈∶0.4%甲酸水溶液(pH3.2)为15∶85]涡旋3 min,LC-MS/MS进样分析。
1.7 方法学验证
在测定条件下,待测物克林霉素和内标保留时间均约为0.3 min,克林霉素和内标通道出峰保留时间处相互未见明显干扰,高浓度进样后未见明显残留。
空白血浆制备标准曲线浓度为:0.02、0.04、0.1、0.5、1.5、9、10 mg/L,进行前处理后进样检测得到克林霉素峰面积与内标峰面积之比和浓度线性回归方程y=0.002 3x+ 0.002 4,R2=0.997 0。线性范围为0.02~10 mg/L,定量下限为0.02 mg/L。
采用定量下限(LLOQ)、低(QCL)、中(QCM)、高(QCH)4个浓度血浆质控样品(0.02、0.06、2、8 mg/L),每个浓度平行6份,批内准确度偏差为-8.5%~5.00%,批内变异系数≤6.99%。批间准确度偏差为-2.50%~0.75%,批间变异系数≤7.08%。
采用QCL、QCM、QCH血浆质控样品,提取回收率分别为77.49%、83.03%、78.33%,变异系数为3.75%。内标平均提取回收率为82.43%,变异系数为4.45%。
QCL、QCH浓度质控样品内标归一化基质效应因子变异系数为5.85%、4.69%。脂血、溶血基质对待测物和内标测定无显著影响。
待测物贮备液和工作液在-20 ℃条件下稳定59 d,在室温避光条件下分别稳定22 h、21 h;内标工作液在室温避光条件下稳定18 h;全血在黄光灯或日光灯下、室温和冰浴条件下均稳定2 h;血浆基质样品室温下稳定24 h,4 ℃条件下稳定48 h,-20 ℃和-70 ℃条件下稳定59 d;前处理后的样品在4 ℃条件下稳定193 h;血浆基质样品在-20 ℃和-70 ℃条件下反复冻融5次后稳定。
方法学验证及生物样本测定由江苏万略医药科技有限公司完成。
1.8 药动学参数计算及生物等效性判定
使用非房室模型方法,采用WinNonlin®6.4版软件,计算药动学参数,对主要药动学参数(Cmax、AUC0-t、AUC0-∞)进行自然对数转换后,考虑周期效应、制剂效应、序列效应、受试者(随序列)效应和残差,以lnCmax、lnAUC0-t、lnAUC0-∞为因变量进行多因素方差分析,对Tmax进行非参数检验。以方差分析结果为基础,采用90%CI分析法,当受试制剂与参比制剂药动学参数AUC0-t、AUC0-∞和Cmax的几何均值比的90%CI均在80.00%~125.00%等效区间内,则认为两制剂生物等效。
2 结果
2.1 受试者入组和完成情况
空腹组受试者和餐后组受试者全部完成试验,均纳入安全性分析集、药动学参数集及生物等效性分析集。空腹给药组共入组24名受试者,男14名,女10名,平均年龄为(26.2±3.6)岁,平均体重为(63.76±9.87)kg,平均体重指数(BMI)为(22.75±2.08)kg/m2,汉族21名,其他民族3名。餐后给药组共入组24名受试者,男14名,女10名,平均年龄为(25.3±5.5)岁,平均体重为(60.32±7.56)kg,平均BMI为(22.17±1.92)kg/m2,汉族21名,其他民族3名。
2.2 血药浓度-时间曲线
空腹组口服克林霉素受试制剂和参比制剂后平均血浆药物浓度-时间曲线见图1,餐后组口服受试制剂和参比制剂后平均血浆药物浓度-时间曲线见图2。无论空腹或餐后给药,服用受试制剂和参比制剂后药时曲线均相似。
2.3 药动学参数
空腹和餐后口服克林霉素受试制剂和参比制剂后主要药动学参数见表1。空腹组参比制剂的Cmax、Tmax、AUC0-t、AUC0-∞分别为3.64 mg/ L、0.75 h、12.70 h·mg/L、13.10 h·mg/L,受试制剂分别为3.55 mg/L、0.62 h、12.20 h·mg/ L、12.40 h·mg/ L。餐后组参比制剂的Cmax、Tmax、AUC0-t、AUC0-∞分别为3.37 mg/L、2.00 h、15.20 h·mg/L、15.70 h·mg/L,受试制剂分别为3.37 mg/L、2.00 h、16.40 h·mg/L、17.00 h·mg/ L。
2.4 生物等效性评价
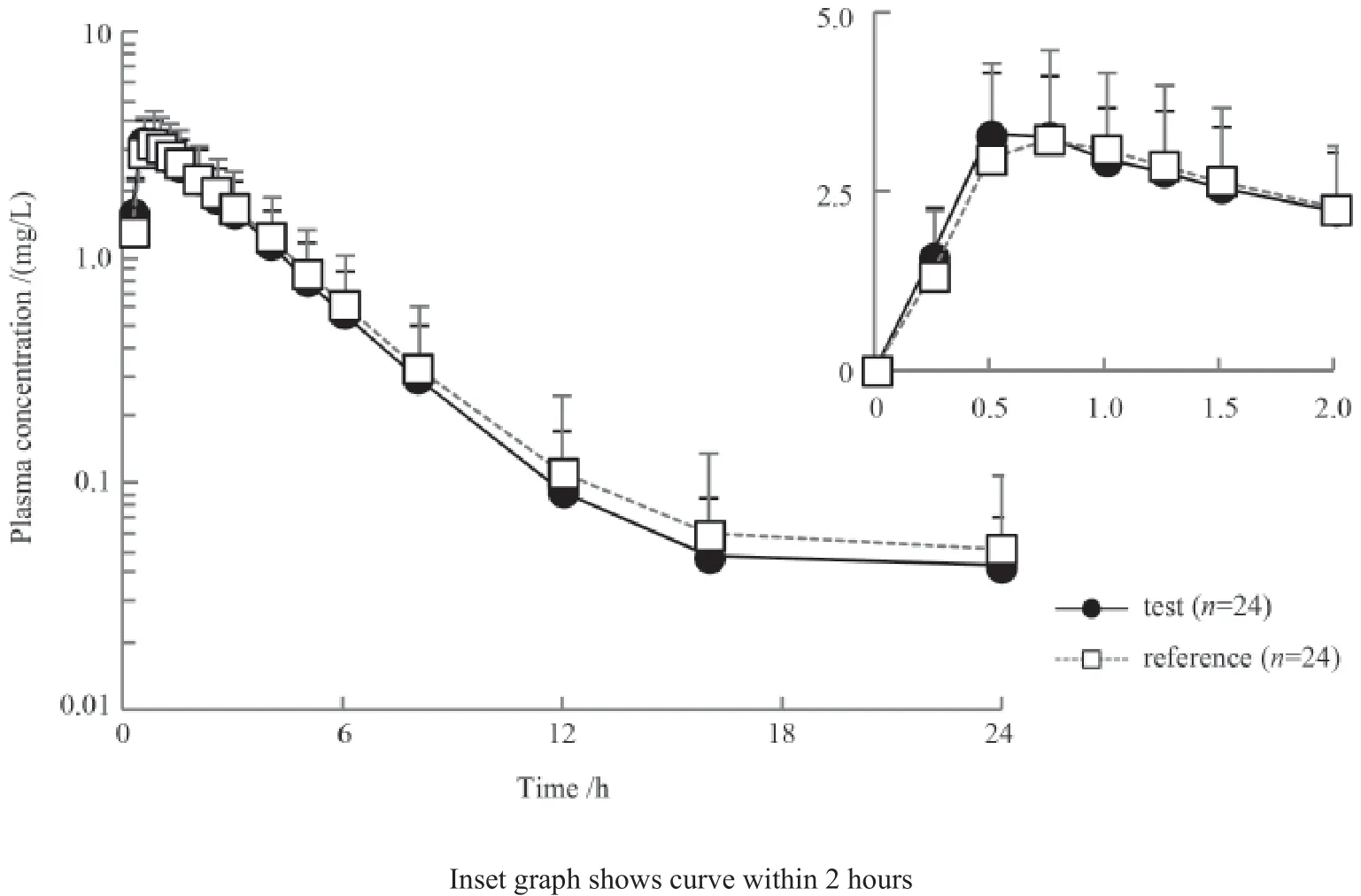
图1 受试者空腹口服克林霉素后平均血浆药物浓度-时间曲线Figure 1 Plasma concentration-time curves of clindamycin in subjects following oral administration under fasted state

图2 受试者高脂餐条件下口服克林霉素后平均血浆药物浓度-时间曲线Figure 2 Plasma concentration-time curves of clindamycin in subjects following oral administration under fed state

表1 受试者空腹或高脂餐后单次口服克林霉素胶囊受试制剂和参比制剂的主要药动学参数Table 1 Mean pharmacokinetic parameters of clindamycin in healthy subjects after single dose administration of test and reference preparations under fasted or fed condition(mean ± SD)
健康受试者空腹口服克林霉素受试制剂和参比制剂后,主要药动学参数Cmax、AUC0-∞、AUC0-t经对数转换后几何均值比分别为99.34%、97.84%、98.47%,进行方差分析和双单侧t检验,90%CI分别为91.94%~107.34%、90.60%~105.64%、91.28%~106.22%。经非参数秩和检验,Tmax差异无统计学意义(P> 0.05)。健康受试者高脂餐后口服受试制剂和参比制剂,主要药动学参数Cmax、AUC0-∞、AUC0-t经对数转换后几何均值比分别为99.87%、106.28%、105.70%,进行方差分析和双单侧t检验,90%CI分别为91.53%~108.97%、98.28%~114.92%、97.81%~114.22%。经非参数秩和检验,Tmax差异无统计学意义(P>0.05)。中国健康受试者空腹或餐后口服受试制剂和参比制剂后,Cmax、AUC0-∞、AUC0-t的几何均值比90%CI均在80.00%~125.00%,因此两制剂生物等效。
2.5 安全性评价
空腹和餐后给药组所有受试者均进入安全性分析集。空腹服用参比制剂的24名受试者中7名发生了11例次不良事件,其中3例次不良事件与参比制剂可能有关,包括荨麻疹、丙氨酸转氨酶和天冬氨酸转氨酶升高。空腹服用受试制剂的24名受试者中8名发生了14例次不良事件,其中3例次不良事件与受试制剂可能有关,包括荨麻疹、呕吐和腹泻。1例发生荨麻疹不良事件的受试者采用炉甘石硫洗剂治疗后恢复。3例次与受试制剂可能有关的不良事件转归均为恢复。餐后服用参比制剂的24名受试者中10名发生18例次不良事件,其中7例次不良事件与参比制剂可能有关,包括血白细胞计数降低、中性粒细胞绝对值降低、中性粒细胞百分比降低、淋巴细胞百分比升高、头晕和头昏。餐后服用受试制剂的24名受试者中5名发生了7例次不良事件,其中2例次不良事件与受试制剂可能有关,包括头晕和中性粒细胞绝对值降低。所有不良事件均为轻度、一过性,除1名受试者发生荨麻疹,使用炉甘石硫洗剂治疗后恢复外均未经治疗自行恢复。试验中未发生严重不良事件,未发生导致退出的不良事件。
3 讨论
克林霉素是临床常用抗生素,抗菌谱与林可霉素相同,但抗菌活性强于林可霉素,口服吸收快速完全,组织穿透性良好,临床上广泛用于敏感革兰阳性球菌和厌氧菌引起的感染。本研究中中国健康受试者单次空腹口服盐酸克林霉素胶囊300 mg受试制剂和300 mg参比制剂后快速吸收,受试制剂和参比制剂Tmax中位数为0.62 h(0.50~1.51 h)、0.75 h(0.50~2.50 h),平均半衰期分别为3.09 h和4.06 h;餐后口服受试制剂和参比制剂后Tmax中位数为2.00 h(0.75~5.00 h)、2.00 h(0.75~4.00 h),平均半衰期分别为5.20 h和3.72 h。进餐使口服吸收略延缓,药物暴露量略增加(AUC增加约20%~30%),Cmax无影响。健康受试者服用受试制剂和参比制剂后发生的与研究药物相关的不良事件发生率相似,主要包括呕吐、腹泻、荨麻疹和轻度、一过性的肝酶异常和血常规异常,所有不良事件均未超出说明书。
对于骨髓炎成年患者的临床治疗方案为600 mg,1次/ 8 h口服或静脉给药。由于体外甲氧西林耐药金黄色葡萄球菌最低抑菌浓度(MIC)为0.125 mg/L[7],文献报道克林霉素骨穿透率约为30%[8],克林霉素骨浓度与MIC之比达到5时可预期达到良好微生物学疗效[9]。因此血浆最低浓度达到2 mg/ L预期可达到良好微生物学疗效。根据本研究中300 mg剂量下暴露量,推测600 mg,1 次 /8 h口服给药时血药浓度接近50%时间将持续高于2 mg/L,预期可达良好疗效。
两制剂在餐后和空腹给药后药时曲线非常相似,发生的不良事件及发生率均相似。经生物等效性评价,两制剂生物等效。
猜你喜欢
今日畜牧兽医(2022年10期)2022-12-23
中国药学药品知识仓库(2022年5期)2022-04-11
浙江化工(2022年1期)2022-02-19
口腔护理用品工业(2021年4期)2021-11-02
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
儿童故事画报·发现号趣味百科(2019年5期)2019-07-14
中国科技纵横(2019年23期)2019-02-14
小天使·五年级语数英综合(2017年8期)2017-08-09
语文世界(小学版)(2017年1期)2017-03-15
中国中药杂志(2016年22期)2017-02-13
