
调节阿片受体MOR胞内信号转导的分子机制研究进展*
2019-10-25 02:50聂登云吕志刚
中国疼痛医学杂志 2019年10期
聂登云 吕志刚△
(1南京中医药大学药学院,南京 210046;2针药结合教育部重点实验室,江苏省针灸学重点实验室,南京 210046)
阿片类药物作为临床上治疗疼痛的主要药物,其广泛应用受到众多不良反应的限制。如何优化阿片类药物的治疗效果,完善其治疗方案已成为临床上治疗疼痛的一个亟待解决的问题。阿片受体的胞内信号转导机制已能够被系统地阐述,其发挥镇痛及不良反应的机制似乎并不完全相同,因此有望找到一条特异性的镇痛信号转导通路而避免不良反应的产生。通过总结近年来的国内外文献发现μ阿片受体基因OPRM1剪接异构体和相关性离子通道的研究日益火热,为改善阿片类药物镇痛效果且减少不良反应提供了新的可能。
G蛋白偶联受体(G protein-coupled receptor,GPCR)是目前发现的最大的一组药效团受体,也是大多数药物作用的靶点。GPCR涉及的病生理功能包括疼痛、癌症、心血管、胃肠、视觉和呼吸以及中枢神经系统,其中包括μ阿片受体(μ-opioid receptor, MOR)、δ 阿片受体 (δ-opioid receptor, DOR)、κ阿片受体 (κ-opioid receptor, KOR)。这些阿片受体的组织分布和内源性配体选择性的不同,可以差异性地调节阿片受体激活的生理效应(见表1)。MOR是一类对于脑啡肽和β-内啡肽具有高亲和性,但对于强啡肽只有低亲和性的阿片受体。DOR一般以脑啡肽为内源性配体,其可能负责调节慢性疼痛的伤害感受。KOR主要以强啡肽为内源性配体,与阿片类药物成瘾性的产生密切相关。虽然DOR和KOR也与阿片类药物的镇痛作用相关,但阿片类药物的镇痛作用主要通过MOR介导[1]。
MOR的镇痛作用主要通过G蛋白跨膜运输,G蛋白通常是指细胞膜上的G蛋白异源三聚体,这种蛋白质由α、β、γ三种亚基构成,后两种被称为β-γ复合物。目前,已经在哺乳动物中发现了16个不同的α亚基家族成员,5个不同的β亚基家族成员和11个不同的γ亚基家族成员[2]。
当配体结合MOR时,诱导受体跨膜结构域发生构象变化,激活G蛋白[3]。这些构象变化改变了G蛋白异源三聚体的相互作用,从而促进了GDP与Gα亚基的解离,之后GTP(在细胞溶质中,GTP的浓度远远高于GDP)与Gα亚基快速结合,引起Gα亚基的三个螺旋“转换区”的构象发生重大变化,由GTP结合诱导的这些区域的构象变化引起Gβ-γ二聚体与Gα亚基的解离,这两者都有生物活性以触发下游信号的转导。G蛋白异源三聚体的解离会激活细胞内调节神经元活性的效应酶,内源性GαGTP酶可以水解GTP上的第三个磷酸基团使其变为GDP失活。最后,Gα亚基与Gβ-γ二聚体重新结合,回到基础状态(见图1)。受体可以激活G蛋白的特定亚型,每种G蛋白都通过调节第二信使的水平或直接调节离子通道功能来介导神经元对神经递质信号的转导。MOR通过腺苷酸环化酶抑制G蛋白家族(G i/o)发出信号,其通过激活G蛋白偶联内流钾通道,并抑制电压门控钙通道来介导下游信号的转导。

表1 不同阿片受体亚型的特点
一、调节MOR信号转导的蛋白质
与其他GPCR一样,MOR的信号转导主要受G蛋白信号转导调节蛋白(Regulators of G protein signaling, RGS)、G蛋白偶联受体激酶(G protein-coupled receptor kinase, GRK)、β-抑制蛋白 (βarrestin)等影响。
1.G蛋白信号转导调节蛋白
RGS是MOR信号转导中最重要的调节蛋白。RGS中由120个氨基酸组成的结构域可以作用于与GTP结合的Gα亚基,加快其GTP的水解速率。哺乳动物RGS家族由大约四十种蛋白质组成,分为九个亚科[4]。许多体内和体外的研究表明,RGS蛋白在MOR中起调节作用。RGS9-2蛋白(属于RGS蛋白的C/R7家族)似乎有效地调节MOR信号转导。RGS9基因敲除小鼠模型的研究表明,RGS9-2蛋白对阿片类药物镇痛作用的敏感性起关键作用。免疫共沉淀实验表明,吗啡处理可促进RGS9-2、MOR和β-arrestin-2之间复合物的形成。此外,RGS9-2可以作为MOR受体内吞的负调节剂,并阻止阿片样物质诱导的ERK1/2磷酸化。RGS9-2复合物可降低吗啡镇痛效应,促进吗啡耐受性的发展;相反地,RGS9-2复合物正向调节其他阿片类药物的镇痛作用,如芬太尼和美沙酮[5]。RGS蛋白正向调节MOR与GRK的偶联,这种偶联对于阿片类药物的脊髓水平的镇痛效应起关键作用[6]。这些研究表明RGS在调节MOR镇痛中起重要作用。

图1 MOR信号转导通路图
2.G蛋白偶联受体激酶
GRK和β-arrestin的相互作用在减弱MOR受体信号转导中起关键作用,它们可以使受体在激动剂的长时间刺激下处于脱敏状态。GRK通过MOR受体C-末端尾部的磷酸化促进β-arrestin与MOR的结合,这阻止了后者与G蛋白异源三聚体的关联,导致G蛋白介导的信号转导的破坏[7]。对于这种调节机制,主要涉及两个蛋白质家族:GRK和第二信使激酶如蛋白激酶A(protein kinase A,PKA)或蛋白激酶C (protein kinase C, PKC)[8]。迄今为止,已经在人类中鉴定了七种GRK,在功能上分为三类:GRK1型、GRK2型和GRK4型[9]。GRK1和GRK7属于GRK1型,主要存在于视网膜中并调节光受体即视蛋白的功能。GRK2型包括GRK2和GRK3,它们都广泛表达并受G蛋白βγ亚基的调控。GRK4型包括GRK4、GRK5和GRK6,其中GRK4具有有限的组织分布,它主要存在于睾丸中,而GRK5和GRK6也广泛表达却对G蛋白βγ亚基不敏感。GRK2在大脑中广泛表达,对神经系统有重要影响。许多实验表明GRK2可以调节MOR的功能发挥,在长期注射吗啡的大鼠的蓝斑区和皮质中,GRK2表达水平有所上升。β-arrestin的募集需要激活GRK2和MOR磷酸化,而GRK募集也依赖于MOR受体C-末端尾部的磷酸化位点[10]。尽管这些研究表明MOR与GRK之间表达水平发生了相关性改变,但尚不清楚这些变化的功能意义。
3.β-抑制蛋白
β-arrestin不光参与了与GRK相关的MOR受体磷酸化的脱敏反应,还介导了MOR受体内吞以减弱G蛋白信号转导。哺乳动物表达出四种arrestin亚型,分别为arrestin-1、arrestin-2(又被称为β-arrestin-1)、arrestin-3(又被称为 β-arrestin-2)和arrestin-4。在MOR信号转导的调节中,主要是β-arrestin-1和β-arrestin-2发挥作用。不同的阿片受体亚型结合的β-arrestin-1和β-arrestin-2不同,其可能产生不同的信号转导机制。在使用GFP标记β-arrestin-2的HEK-293细胞或使用显性阴性βarrestin-2的纹状体神经元实验中发现,激动剂依赖性激活的MOR可以募集β-arrestin-2[11]。敲除βarrestin-2的小鼠表现出吗啡镇痛效应的增强和镇痛持续时间的延长,这表明β-arrestin-2在调节MOR功能中的重要性。除了参与减弱G蛋白介导的信号转导,研究还表明β-arrestin可以诱导细胞外调节蛋白激酶(extracellular regulated protein kinase, ERK)持续性磷酸化,这与G蛋白诱导的ERK瞬时磷酸化不同[12]。在加压素1b受体(vasopressin 1b receptor, V1bR) -μ受体复合物中,V1bR C-末端中富含亮氨酸的片段对于β-arrestin-2的结合是必需的,缺失这种富含亮氨酸的片段增强了吗啡的镇痛效果及降低了吗啡的耐受性[13]。总之,这些研究表明βarrestin作为G蛋白依赖性信号转导机制与独立性信号转导机制之间的桥梁,在介导MOR信号转导中发挥重要作用。
4.其他蛋白质
能影响MOR信号转导的一种蛋白质是钙调蛋白(calmodulin, CaM),CaM是一种普遍存在的Ca2+敏感性调节蛋白,其参与调节包括腺苷酸环化酶、Ca2+/CaM依赖性激酶和磷酸酶、离子通道等多种细胞质酶。有研究表明,支架蛋白spinophilin可与MOR发生相互作用,spinophilin是参与调节MOR信号转导和受体内吞作用的信号复合物的一部分,spinophilin基因敲除小鼠对吗啡镇痛的敏感性降低以及更快地产生耐受性[14]。与MOR相互作用的另一种支架蛋白是Tamalin。在伏隔核中,Tamalin似乎是信号复合物的一部分,通过与MOR结合调节吗啡的镇痛作用[15]。总之,这些研究表明蛋白质-蛋白质的相互作用在调节MOR信号转导中起重要作用。
受体内吞是激动剂激活的阿片受体复原和再敏化所必需的。许多研究表明,MOR的受体内吞需要β-arrestin募集。涉及MOR内吞作用的另一种蛋白质是p38丝裂原活化蛋白激酶(p38mitogen-activated protein kinase, p38 MAPK),即使在没有激动剂的情况下,p38MAPK的磷酸化足以引起MOR的自源性受体内吞。
如今多项研究表明不同的阿片类药物诱导产生不同的蛋白质复合物,蛋白质复合物的形成差异将导致阿片受体之间药物靶向的差异[16]。MOR是组成型活性的,可在不存在配体的情况下激活G蛋白。在DRG神经元中,实验证实了组成型活性的MOR可以被有效地从细胞表面去除,该过程似乎依赖于β-arrestin-2[17]。
蛋白质与蛋白质的相互作用和受体与蛋白质的相互作用是所有细胞功能的核心,其对细胞内外刺激反应的动态变化调节着细胞的信号转导。MOR的信号转导可以通过与细胞骨架蛋白、信号分子、酶、激酶等多种蛋白质的相互作用来调节,这也是MOR发挥镇痛作用的关键因素。研究表明阿片类药物产生的镇痛作用是通过MOR中G蛋白的信号转导而实现的,而包括呼吸抑制、便秘和耐受[18]等的许多不良反应,可能是通过活化MOR下游的β-arrestin途径信号转导的。
二、MOR剪接异构体
μ阿片受体基因(μ-opioid receptor gene, OPRM1)的选择性剪接是广泛存在的[19],啮齿动物和人类都可以产生具有不同结构类型的剪接异构体,根据跨膜结构类型分为3类:全长7次跨膜(7 transmembrane, 7TM)剪接异构体、缺少外显子1的截短型6TM剪接异构体和仅包含外显子1的截短型1TM剪接异构体[20]。OPRM1的选择性3'剪接可产生独特的C-末端氨基酸序列,导致C-末端中存在广泛的潜在性磷酸化位点,其可以通过调节β-arrestin的功能来发挥MOR的不同作用。除了C-末端氨基酸序列,这些7TM剪接异构体的其余部分是相同的,因此它们具有相同的结合位点。迄今为止的研究表明,小鼠、大鼠和人类的OPRM1基因分别可产生29,16和19种剪接异构体[21],其中大多数是具有7TM全长结构域的剪接异构体(见图2)。截短型6TM剪接异构体在药理学上是十分重要的,尤其是在几种常用的阿片类镇痛药中起主要作用,为研发新的阿片类镇痛药物提供了靶标[22]。截短型1TM剪接异构体不直接结合阿片类药物,它们主要通过伴侣蛋白的作用增强全长7TM剪接异构体的表达,从而有助于发挥阿片类药物的镇痛作用[23]。
MOR剪接异构体的选择性激动可以诱导产生不同的行为效应:在129S7/SvEv和C57BL/6J背景下mE3M和mE4M纯合子小鼠中的DAMGO和吗啡诱导的刺激水平显著降低,但在mE7M-129和mE7M-B6纯合子中没有显著降低。在热辐射甩尾实验中,吗啡镇痛对C-末端截短的6TM剪接异构体小鼠没有太大影响。在mE3M-B6和mE4M-B6纯合子小鼠中,吗啡耐受性会更快且更大程度地发展,这种耐受性的增强在mE3M-129和mE4M-129纯合子小鼠中更为明显。与WT-B6对照小鼠相比,mE3M-B6和mE4M-B6纯合子小鼠的纳洛酮诱导的沉降跳跃显著减少。mE3M-B6和mE4M-B6纯合子小鼠中吗啡诱导的条件性位置偏好(conditioned place preference, CPP)与它们各自WT对照小鼠所见的相似;相反地,mE7M-B6纯合子小鼠的CPP显著低于mE7M-WT对照小鼠,表明外显子7关联的剪接异构体参与C57BL/6J小鼠的吗啡奖赏行为。与WT-B6对照小鼠相似,mE7M-B6纯合子小鼠在吗啡处理后显示出丘脑中ERK活化增加而其他区域没有变化,表明与外显子7关联的剪接异构体C-末端序列可能不参与ERK1/2的激活[20]。[35S]GTPγS受体结合实验作为评价受体与G蛋白脱偶联的关键检测方法,其测定剪接异构体C-末端在结合效能标记中的不同显示,揭示了C-末端远端序列的差异可以影响MOR激动剂诱导的受体-G蛋白偶联和信号转导。这些MOR剪接异构体具有不同的分布、转导和脱敏特性。有实验表明,大鼠的MOR剪接异构体C-末端可调节激动剂诱导的受体内吞和再敏化。吕志刚教授等使用QPCR技术测定了 C56BL/6J、129P3/J、SJL/J、SWR/J近交系小鼠中前额皮质、纹状体、丘脑、下丘脑、海马、脑干、中脑导水管周围灰质、小脑和从L1到L5脊髓中OPRM1不同剪接异构体mRNA 的表达水平,发现长期吗啡处理可选择性地使大脑不同区域中C-末端剪接异构体mRNA表达水平增加多达300倍[24]。几种吗啡诱导行为和受体脱敏的相似性表明与外显子7关联的剪接异构体C-末端的功能可能与β-arrestin-2相关[20]。6TM剪接异构体与β-arrestin-2的共表达和二聚化可以使6TM剪接异构体从细胞内区室转移至质膜,其形成的异源二聚体成为阿片类药物产生兴奋性和痛觉过敏的六次跨膜信号转导机制的基础[25]。最新的研究证明了6TM剪接异构体在一种新 型 MOR 激 动 剂 (3-iodobenzoyl-6β-naltrexamide,IBNtxA)镇痛中的药理学重要性:6TM剪接异构体对于阿片类药物发挥镇痛作用是必需的[26],并证实了与MOR跨膜结构域对应的外显子2和3编码的共同功能核心足以激活其活性[27]。在不同的MOR激动剂中观察到的体内效应的差异被归因于与不同的MOR剪接异构体相互作用的变化;然而在体内,激动剂对于剪接异构体是否具有足够的选择性,或者选择性剪接异构体的表达是否适用于鉴别奖赏、镇痛和耐受等的过程还没有被明确证明[16]。
三、调节MOR的离子通道
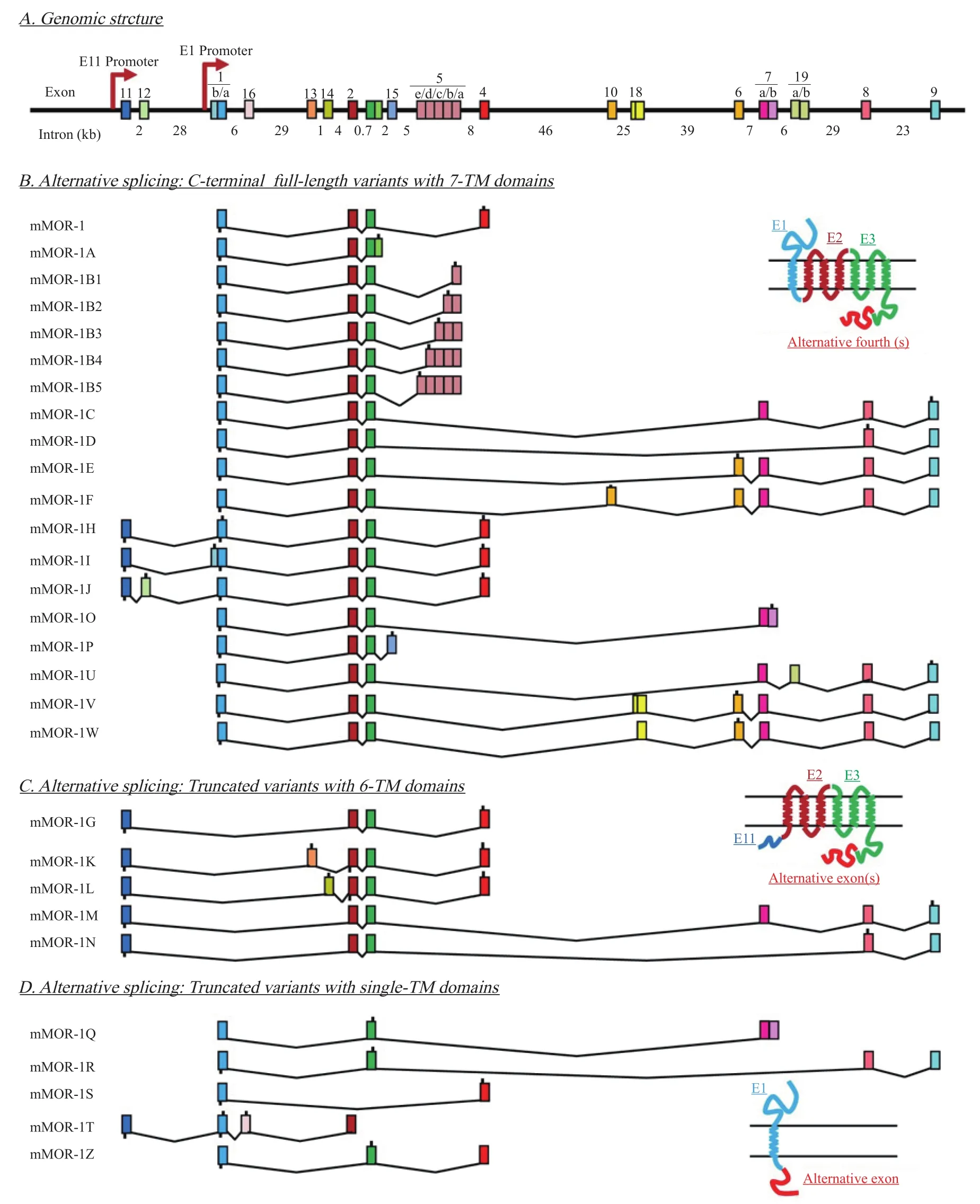
图2 小鼠OPRM1基因结构、可变剪接和预测的蛋白质结构的示意图[21]
MOR和离子通道可将外周伤害感受器的有害刺激转换成电信号。有害刺激的检测器中最常见的一组是瞬时受体电位通道(transient receptor potential cation channel, TRP)家族,主要包括TRPV1和TRPA1。这些疼痛性离子通道表现出伤害感受器的功能特性,其在神经元中选择性表达从而可能减少特定药物对它们的不良反应。外周炎症产生的多种炎症介质可以作用于由伤害感受器表达的同源受体以激活胞内信号转导通路,这些通路可以磷酸化TRP通道,从而改变它们向细胞膜运输离子的动力学阈值。痛觉神经元中TRPA1和TRPV1的钙激活可作为信号放大器以增加细胞内钙的浓度。TRPV1是一种非选择性阳离子渗透通道,对钙的选择性更高,具有4倍对称性的均四聚体形式,每个单体由六个跨膜螺旋和一个孔环片段组成。阿片类药物与TRPV1不是直接的相互作用,主要通过蛋白激酶如PKA和PKC等激活TRPV1[28]。实验表明TRPV1的激活通过钙流入导致CaM依赖性的GRK5远离细胞膜,从而阻断磷酸化MOR1并阻止MOR1内吞。TRPV1结合MOR1可以阻断MOR1的阿片类物质依赖的磷酸化,同时保持G蛋白信号转导过程的完整[29]。TRPV1可能通过伏隔核中的p38MAPK信号通路来调节吗啡诱导的条件性奖赏效应[30]。还有研究表明,TRPV1的拮抗剂可减少小鼠切口手术后阿片类药物需求量[31],外周性MOR的激活能减弱TRPM3依赖性疼痛[32]。这些研究都展示了操纵特定离子通道来治疗疼痛,也许是镇痛领域一个新颖且很有前景的方向。
四、MOR与外周免疫细胞来源的阿片肽
阿片类药物发挥的镇痛作用也可能受到炎症期间来自特定免疫细胞的阿片肽、β-内啡肽、甲硫氨酸脑啡肽和强啡肽等外周释放的影响,这些是粒细胞、淋巴细胞和单核细胞等在炎症过程中不同阶段释放的阿片类物质[16]。这些阿片类物质与初级感觉神经末梢上的MOR相结合,启动阿片类物质信号级联以增加超极化,抑制神经递质的释放,从而减轻疼痛的感觉[33]。从外周免疫细胞中释放的阿片类物质似乎不会引起耐受性,并且与阿片类物质的中枢性不良反应无关[34]。这表明局限于外周的阿片类药物可能比中枢性阿片类药物更适用于治疗外周疼痛。
五、MOR的晶体结构
MOR的晶体结构展现了标准的7个跨膜结构域,记录了不同激动剂结合位点的构象,例如DAMGO,BU72[35]和PZM21[36]。所有共享外显子1、2、3的全长7TM剪接异构体都具有相同的结合位点,以相似的亲和性与不同的阿片类药物结合,但发挥的镇痛效应却不尽相同,这可能与MOR的偏向信号转导有关。有研究证明了TRV130作为一种新型的G蛋白偏向阿片类药物具有更好的镇痛效果而减少了不良反应[37]。相信随着MOR晶体结构的进一步阐释,我们可以发现更多的G蛋白偏向型阿片类药物来优化其镇痛效果。
六、结语
目前临床上使用阿片类药物,其不良反应仍然无法避免,但必须认识到阿片类药物在治疗疼痛方面具有重要的价值。不同的MOR激动剂可以通过不同的机制诱导MOR脱敏,G蛋白信号转导的是镇痛作用,而GRK和β-arrestin的信号转导会产生阿片类药物的不良反应等,这些表明可以通过找到特异性的G蛋白信号转导通路而避免β-arrestin引起的受体磷酸化来优化阿片类药物的治疗效果,可是至今以来的研究并未发现有如此特异性的G蛋白信号转导通路,而更多的研究关注于如何减少GRK和β-arrestin的信号转导但进展缓慢。希望通过对MOR剪接异构体、离子通道和晶体结构的进一步研究,能够发现一种新的阿片类物质镇痛机制来指导治疗疼痛相关性疾病,同时有效地降低甚至完全消除阿片类药物的不良反应。
猜你喜欢
临床与实验病理学杂志(2021年7期)2021-09-06
湖南农业大学学报(自然科学版)(2021年4期)2021-08-14
云南医药(2021年3期)2021-07-21
特别健康·下半月(2020年3期)2020-03-18
智富时代(2019年6期)2019-07-24
智富时代(2019年6期)2019-07-24
物理学报(2019年12期)2019-06-29
江苏农业科学(2017年14期)2017-10-10
西部学刊(2017年8期)2017-09-08
职工法律天地·下半月(2016年4期)2017-05-31
