
苹果SLAF图谱构建及果锈基因QTL分析
2019-11-05 10:03张朝红陈东玫杨凤秋赵同生赵国栋赵永波
华北农学报 2019年5期
张朝红,陈东玫,杨凤秋,赵同生,李 扬,赵国栋,赵永波
(河北省农林科学院 昌黎果树研究所,河北 昌黎 066600)
果锈(Russeting,RUS)是苹果[1]、梨[2]、葡萄[3]、马铃薯[4-5]等作物的重要性状之一。马铃薯的锈变品种Russet Burbank因综合了高产、低糖、吸油少、耐贮藏、适宜加工等特性,在美国、加拿大、法国、英国、新西兰等多地广泛种植[4,6]。梨的果锈与果皮色泽密切相关,并对生物、非生物胁迫起到一定的保护作用[2,7]。苹果多锈品种金冠、Elstar、桔苹因果锈的频繁发生使其果实的商品价值锐减[8-11],成为限制该类品种持续发展的一大障碍。
苹果、梨等树种幼嫩果实(或块茎)受到气候、病菌、药剂等因子的作用时,它的表皮细胞异常分裂、增殖而开裂,其下层细胞膨大木栓化产生木栓形成层,进而形成木栓组织,此时表皮外层的角质层不断龟裂剥落,在果实(或块茎)表面形成了黄褐色、赤褐色周皮组织即果锈[8-9,12-17]。果锈的发生,在幼果期(金冠苹果,花后2周)即开始,此后的2~6周是果锈形成的关键时期[9,17]。从生理学角度分析,苯丙烷类代谢、乙烯代谢及次生代谢等多种途径都与果锈的形成有关,并且木质素生物合成、多胺及过氧化氢的信号传递可对果锈的形成进行调控[18-19]。
果锈基因主要包括表皮物质合成基因和胁迫应答基因2大类,根据功能的不同表皮物质合成基因可进一步分为表皮生物合成基因、木栓质沉积基因。在梨果锈形成过程中,表皮生物合成基因随着胁迫应答基因、木栓质沉积基因的急剧增加而受到抑制[11,20]。在苹果多锈品种中,一些编码纤维素合成酶6、木聚糖合成酶、果胶甲基酯酶的基因表现出了上调表达,与木栓质相关的苯丙烷类代谢调控基因的差异表达,其中的转录因子MdMYB93在木栓质的累积过程中发挥主要作用[11,21]。在砀山酥梨的锈变单株中检测到10个ABC家族的锈变差异表达基因,这些基因参与了锈变果实木栓化的形成[22]。
果锈基因的遗传机制极为复杂。在梨上,果锈基因是受2对基因控制,其中的R1/r1基因的SSR标记CH01c06和Hi20b03,遗传距离分别为 4.8,12.0 cM,推测该基因可能位于梨的LG8上[7],也有学者认为,果锈属于质量性状,而果锈含量则为数量性状,两者分别受不同的遗传位点控制[20]。在苹果上,果锈的遗传力中等,通常在0.34~0.54[23-24];近年来获得了1个控制Renetta Grigia di Torriana锈变的主效基因,它位于苹果的LG12染色体上[25],然而,关于果锈基因的分子遗传机制尚不清楚。2015年在宫崎短枝富士(MYS)×坂田津轻(SKT)的杂种分离群体中发现了全锈的子代单株,通过3 a观察该株子代表现稳定。本研究以该群体的子代植株为试材,采用SLAF技术开发标签并构建图谱,结合表型数据分析,对苹果果锈基因进行遗传定位,以期找到控制苹果果锈的遗传位点,为无锈苹果或免套袋苹果品种的选育及利用提供理论依据。
1 材料和方法
1.1 试验材料
2003年以宫崎短枝富士为母本,坂田津轻为父本杂交,2004年播种杂交种子,2010年转接于SH6中间砧砧木上,进行常规的果园管理。进入结果期后,在该分离群体中发现了全锈苹果变异株(图1)。

1.全果锈苹果(成熟期);2.全果锈苹果(幼果期);3.对照。1. Full russeting apple at ripen stage; 2.Russeting in young fruits;3.Control.
1.2 试验方法
采集宫崎短枝富士×坂田津轻杂种分离群体中的150株子代及双亲的苹果叶片,采用CTAB法分别提取基因组DNA后,运用SLAF-seq 技术(Specific-locus amplified fragment sequencing)和HighMap 软件进行高密度分子标签开发,构建高密度遗传图谱,然后对果锈基因进行QTL定位。
1.2.1 表型调查 参照Durel等[23]的方法,稍有改进,设4级:0级无或没有见到;1级果面有少量果锈(少于1/3),2级果面有大量果锈(介于1/3和1/2之间);3级果面锈量1/2以上。2015-2017年连续3 a对杂种分离后代果面果锈情况进行调查。
1.2.2 酶切方案设计及测序 根据苹果(MalusdomesticaBorkh.)基因组大小以及 GC 含量等信息进行酶切预测。选择RsaⅠ 和HaeⅢ 酶切组合,酶切片段长度在 364~464 bp 的序列定义为 SLAF标签,预测可得到 123 512 个 SLAF 标签。根据最适酶切方案,对检测合格的各样品基因组 DNA 分别进行酶切。得到的酶切片段(SLAF 标签)进行 3′端加 A 处理、连接 Dual-index[26]测序接头、PCR 扩增、纯化、混样、切胶选取目的片段,文库质检合格后用IlluminaHiSeqTM 进行 PE125bp 测序。
1.2.3 SNP开发及编码 根据 Clean Reads 在参考基因组的定位结果,使用 Picard 进行去重复、GATK 进行局部重比对、碱基质量值校正等预处理,以保证检测得到的单核苷酸多态性(SNP)准确性,再使用 GATK 进行SNP的检测,过滤,并得到最终的 SNP 位点集。根据遗传学通用的等位编码规则对多态性标签进行基因型编码。
1.2.4 图谱构建 为保证遗传图谱质量,将多态性 SLAF 标签进行过滤:过滤掉父母本测序深度 4×以下的标签;对于单一多态性标记位点,选择100 个子代中至少有 70 个个体有确定基因型的标记;用偏分离标记处理方法过滤严重偏分离(卡方检验P<0.001)的多态性标记。
将筛选获得的标记,通过计算两两标签之间MLOD 值[27],过滤掉与其中SLAF标签的MLOD值低于 3 的标签,将标签分到 17 个群。以连锁群为单位,采用 HighMap 软件分析将获得连锁群内 Marker 的线性排列,并估算相邻 Marker间的遗传距离。
1.2.5 QTL分析 基于表型数据值,采用mapqtl方法对果锈基因进行QTL分析,LOD阈值为3.0。
2 结果与分析
2.1 测序数据统计与评估
利用Hiseq2500平台进行SLAF-seq文库的测序分析。结果表明,测序获得了 110 321 714 552 bp数据,测序平均 Q30 为 95.07%,平均 GC 含量为 40.11%,样本 GC 分布正常。宫崎短枝富士和坂田津轻分别获得29 944 687 reads (8 972 246 842 bp)、29 251 169 reads(8 763 175 910 bp)数据,测序平均Q30为 93.68%,93.86%,平均GC含量为 38.36%,38.30%;子代群体获得了3 086 176 reads,617 235 279 bp数据,测序平均Q30为 95.09%,平均GC含量为 40.14%;各样本GC分布正常(表1)。

表1 样品测序数量Tab.1 Quantity of sequencing sample
2.2 建库评估
将测序获得的231 821 reads,通过 SOAP软件[28]对 Control 的测序 reads 与参考基因组进行比对。结果表明,Control数据的建库双端比对效率在89.17%,酶切效率为88.41%,SLAF建库正常。
2.3 标记开发
使用 GATK 软件工具包分别对亲本和子代的数据进行比对和SNP开发。结果显示,宫崎短枝富士和坂田津轻分别开发了4 669 811,4 951 676个SNP标签,杂合比率为69.35%和81.65%。子代群体开发了1 116 418个标签,杂合比率为10.83%(表2)。同时,还开发了SLAF 20 440个,平均测序总深度为2 538 482 cM,平均测序深度为12.01 cM。
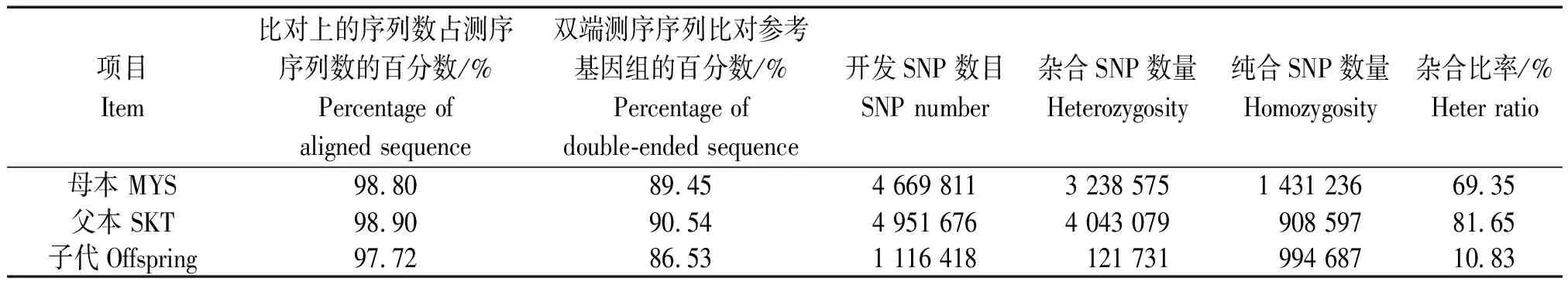
表2 测序数据比对和SNP统计Tab.2 Comparison of sequencing data and SNP
开发的SNP标记中非 aa×bb 标签的数目为 3 989 143个,占总开发的 SNP 数目的 54.50%(表3)。
2.4 遗传图谱构建
过滤筛选后的标记根据MLOD值进行连锁群的划分,所有标签分别归为17个群绘制连锁群。然后以连锁群为单位,采用Highmap将群内标记进行线性排列,共获得上图标记4 075个,总图距为2 235.23 cM(图2)。

表3 SNP的分类统计Tab.3 Statistic of SNP Marker
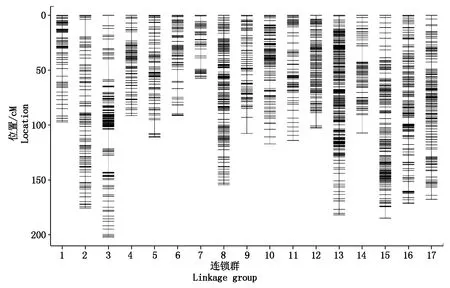
图2 宫崎短枝富士×坂田津轻杂种分离群体的遗传图谱Fig.2 Genetic map constructed with a population derived from Miyazaki Spur×Sakata Tsugaru
2.5 遗传图谱评估
构建的中性遗传图谱,将4 075个标记定位在17个连锁群上,每个连锁群平均包括240个标记,2个标记的平均图距在0.55 cM(表4)。上图标记中包含了256个偏分离标记,占标记总数的比例为6.28%。
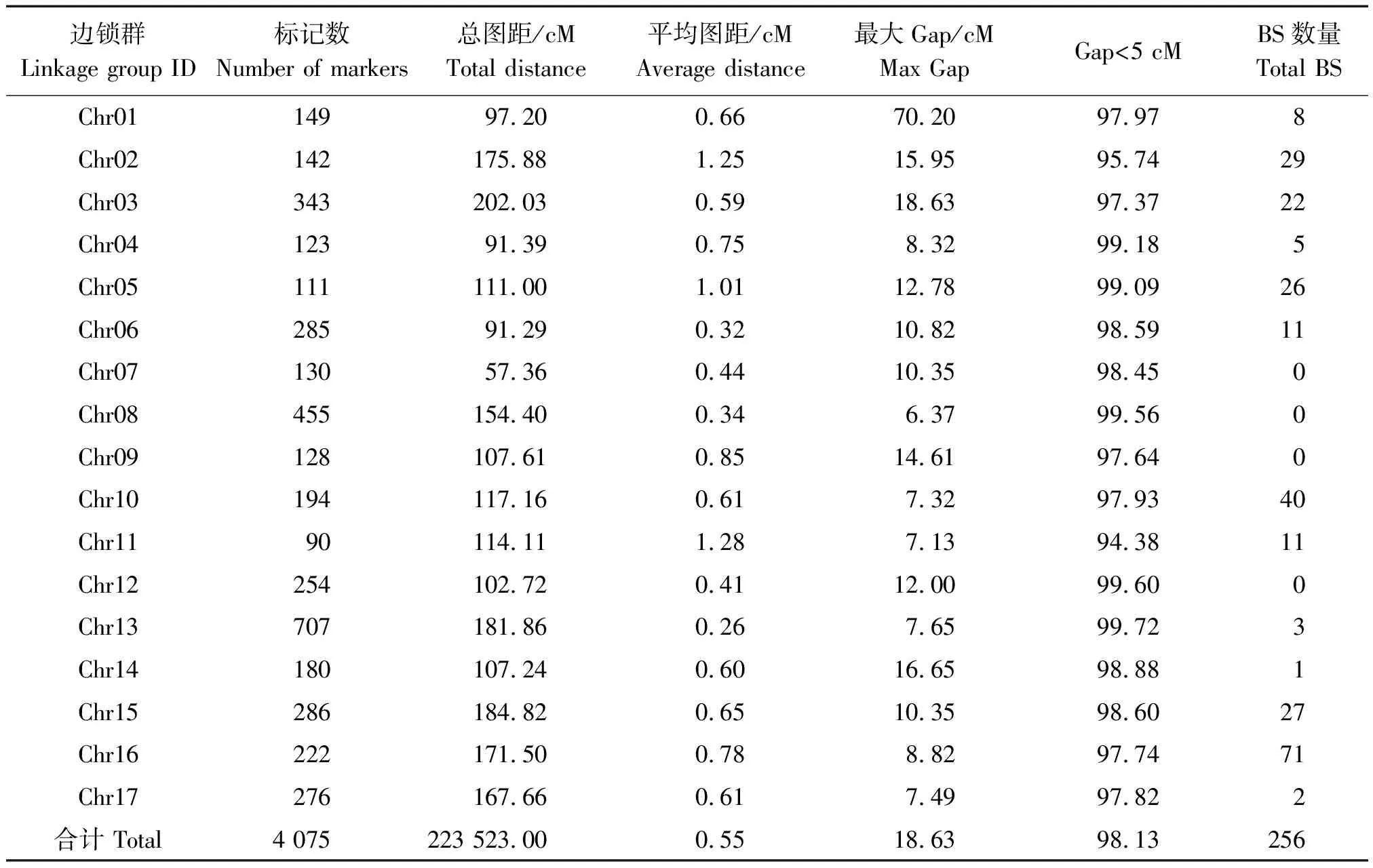
表4 分子标记及偏分离标记位点在连锁群上的分布Tab.4 Basic characteristics of Malus linkage groups in the cross of between Miyazaki Spur and Sakata Tsugaru
对作图群体每个个体上图标记完整性分析发现,上图标记的完整度平均为 99.92%,图谱基因分型准确。将上图标记在基因组上的位置和遗传图谱进行共线性分析发现,各个连锁群上大部分标记的顺序与基因组保持一致,说明共线性较好,遗传重组率的计算准确度高。
2.6 QTL分析
连续3 a的田间调查发现,在调查的150株中,8株无明显果锈,占调查数的5.3%,22株果锈量为1级,占调查数的14.7%,44株果锈量为2级,占调查数的29.3%,还有76株的锈含量在年份间差异较大。经mapqtl分析,共检测到控制苹果果锈的9个QTL,分布在Chr3、Chr9、Chr11和Chr15染色体上(图3),标记距离为0~4.9 cM,分别将这些QTL命名为Qru-3、Qru-9、Qru-11和Qru-15(图4),单个QTL的贡献率为18.0%~85.5%(表5)。其中,Qru-9、Qru-15和Qru-3的贡献率较高(分别为85.5%,24.3%~47.8%和46.1%),可能是控制苹果果锈的关键作用位点。

图3 果锈基因的QTL检测Fig.3 QTL analysis of apple russeting gene
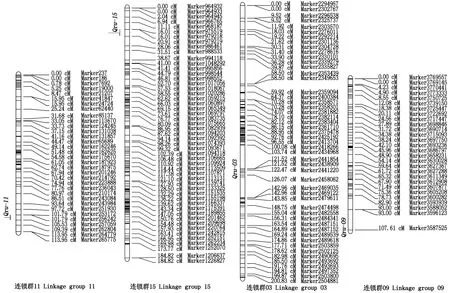
图4 利用宫崎短枝富士和坂田津轻杂种分离群体检测到果锈基因的QTLFig.4 QTL for apple russeting in a population derived from a cross between Miyazaki Spur and Sakata Tsugaru

染色体Linkage group ID位置/cMLocation标记MarkerLOD贡献率/%PVE Chr11101.79Marker2531723.7222.0Chr11101.79Marker2562423.7421.8Chr11105.53Marker2570593.0318.0Chr1516.01Marker9755194.3524.3Chr1516.01Marker9792184.8147.8Chr1516.01Marker9755204.8147.8Chr1520.91Marker9792194.0647.1Chr3123.42Marker24224813.0446.1Chr9107.61Marker35875255.9085.5
对标记区域进行进一步基因注释,注释到127个基因,该4个染色体各包括61,64,1,1个基因。GO注释结果可知,它们在细胞组分、分子功能及生理代谢方面发挥作用,其中Qru-11、Qru-15所携带信息更多一些,是今后研究的重点(表6)。
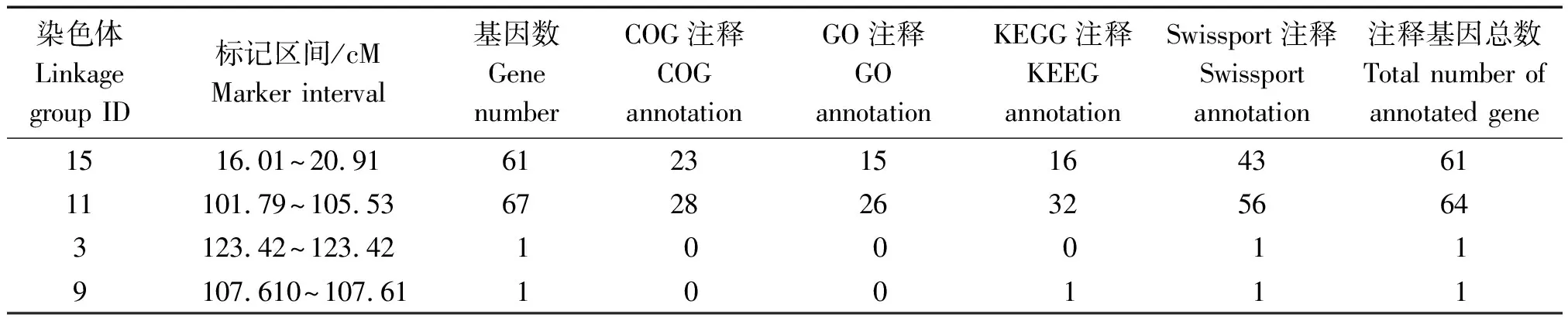
表6 标记区域的基因注释Tab.6 Annotation of the marked regions of apple russeting gene
3 结论与讨论
SLAF-seq技术是利用二代高通量测序发展而来的一套简化基因组测序技术。根据参考基因组信息,通过特定内切酶酶切基因组并筛选特定大小的片段建库测序,从而选择性地得到目标基因组的片段序列。目前,基于 SLAF-seq 的分子标记技术已经在黄瓜[29]、葡萄[30]、木薯[31]、石槲兰[32]、玉米[33-34]等植物中得到应用。本研究利用该技术开发SLAF标签20 440个,SNP标记位点7 309 729个,这些信息位点为苹果相关基因的定位和分子辅助育种研究奠定了基础。
以往研究表明,苹果果锈受气候、病菌等因素的影响,苹果果锈的遗传力中等[23-24],而该基因的大小及所在的位置尚不清楚。近年来,随着高通量测序技术的发展,为性状的遗传分析提供了强大的技术支持[35]。Falginella等[25]将Ru-RGT定位在LG12上400 bp的基因片段上,进一步分析还发现,嘎拉苹果上也拥有该基因位点,但是桔苹等一些多锈品种没有检测到。因此,果锈基因的遗传机制更为复杂,为进一步发掘新的果锈基因,本研究在开发标记的基础上,进行遗传图谱的构建并结合3 a的表型数据,新获得了9个果锈相关QTL,分布于Chr3、Chr9、Chr11和 Chr15上,在这些区段内共注释到127个基因,它们在细胞组分、分子功能及生理代谢方面发挥作用。参照RNA混池重测序技术发现的206个参与细胞构建、生物代谢等过程的差异表达基因[2],进行对比分析、验证,有助于果锈基因的分子调控机制深入揭示。
一直以来,苹果果锈基因被作为不利性状来看待[8-14],不过最近在默顿和皇家嘎拉苹果的果锈组织上检测到桦木酸-3-反式-咖啡酸酯等3种三萜烯-咖啡酸酯,它们都具有独特的免疫调节活性[1],为果锈基因的开发利用提供了新思路。多年来的观察发现,果面锈含量多的果肉更脆,本研究群体中发现的全锈株即是如此,但是果实肉质是与果锈基因连锁,还是因受果锈相关代谢活动的直接影响所致,具体原因有待进一步研究。
猜你喜欢
军事文摘(2022年24期)2022-12-30
分子催化(2022年1期)2022-11-02
北京航空航天大学学报(2022年8期)2022-08-31
军事文摘(2022年16期)2022-08-24
今日农业(2021年11期)2021-08-13
昆明医科大学学报(2021年3期)2021-07-22
烟草科技(2021年6期)2021-06-24
少先队活动(2020年12期)2021-01-14
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
