
云锦杜鹃转录组分析
2019-11-13 02:54许蔷薇楼雄珍林二培童再康
浙江农林大学学报 2019年6期
许蔷薇,楼雄珍,杨 彬,林二培,童再康
(1.浙江农林大学 林业与生物技术学院,浙江 杭州311300;2.浙江农林大学 省部共建亚热带森林培育国家重点实验室,浙江 杭州311300)
随着第2代测序技术的发展,转录组测序(RNA-Seq)已成为植物分子生物学研究的重要手段,广泛应用于功能基因挖掘、分子标记开发、代谢通路和调控机制研究等方面[1-3]。该技术具有成本低、数据量大、效率高、准确性高等优点[4],可对组织或者细胞中所有RNA进行测序,并通过读段(reads)的拼接和丰度统计获得相应的转录本序列信息及其表达水平[5]。云锦杜鹃Rhododendron fortunei为杜鹃花科Ericaceae杜鹃花属Rhododendron植物,为中国特有种,主要分布于安徽、湖南、湖北、浙江等省[6]。云锦杜鹃叶形大,花形美丽具清香,具有较高的园艺观赏价值;适应性较强,易人工栽培,也常作为杜鹃属种间杂交的亲本;此外,枝叶等组织中含有槲皮素、山柰酚、杨梅素等有效活性成分,可开发入药[7-8]。可见,云锦杜鹃作为一种兼具观赏价值和药用价值的优良木本花卉,开发潜力巨大,具有良好的产业化前景。目前,云锦杜鹃的研究主要集中在野生资源调查[9]、群落结构[10]、无性繁殖技术[11]、有效成分分析[8,12]、光合特性[13]、菌根共生等方面[14]。由于缺乏遗传信息,云锦杜鹃的遗传多样性、分子辅助育种等的研究较为滞后。本研究通过RNA-seq高通量测序技术对云锦杜鹃转录组进行测定,通过序列拼接、功能注释和分析,获取大量的序列信息,旨在为云锦杜鹃分子标记开发,遗传多样性分析,以及功能基因挖掘、重要性状形成分子机制等研究提供序列信息。
1 材料与方法
1.1 植物材料
前期以天台华顶林场云锦杜鹃优株茎段为外植体,通过组培获得云锦杜鹃无性系ZL-01。以该无性系组培苗为材料,取其嫩叶、嫩茎和根,液氮冷冻后保存于-80℃用于RNA提取。
1.2 RNA提取与质控
采用Trizol试剂(Invitrogen),按照试剂说明书分别提取组培苗根、茎、叶的总RNA。采用Agilent 2100 Bioanalyzer(Agilent),对总RNA样品的纯度、浓度和完整性进行检测评估,等量混合后用于测序文库构建。
1.3 测序文库构建
取检验合格的RNA,用带有Oligo(dT)的磁珠富集mRNA,将得到的mRNA随机打断成短片段,用六碱基随机引物合成一链cDNA,然后加入缓冲液、 dNTPs、DNA polymerase I和RNase H合成二链cDNA。双链cDNA经纯化后进行末端修复,加ploy(A)并连接测序接头,通过PCR扩增及纯化后得到测序文库。采用Illumina Hiseq 2500进行转录组的测序,测序由北京诺禾致源生物公司完成。
1.4 转录组数据分析
针对测序得到的原始数据(raw data)进行接头和低质量测序片段(reads)去除等处理,获得高质量的干净数据(clean data);计算干净数据的Q20和Q30,GC含量和碱基错误率,评估测序质量;利用序列的重叠,通过Trinity软件对干净数据数据进行序列组装,将短测序片段延伸成较长的片段,并得到片段集合, 利用 De Bruijin方法[15]得到转录本(transcripts)和单基因簇(unigene)序列。
1.5 基因功能注释
利用BLAST和HMMER软件将云锦杜鹃单基因簇序列与公共数据库进行比对,根据基因的相似性进行功能注释,得到与给定单基因簇具有最高序列相似性的蛋白,从而得到该单基因簇的蛋白功能注释信息,其中BLAST参数E≤1e-5,HMMER参数E≤1e-10作为筛选标准。比对采用的公共数据库包括美国生物信息中心(NCBI)非冗余蛋白数据库(Non-redundant protein database,Nr),NCBI核酸序列数据库(Non-redundant nucleotide sequences, Nt), 蛋白质序列数据库(Swiss Prot protein database, SwissProt),真核直系同源基因数据库(Eukaryotic orthologous groups,KOG),蛋白质家族数据库(Protein families database,Pfam),基因本体论数据库(Gene ontology,GO)及京都基因与基金组百科全书(Kyoto encyclopedia of genes and genomes, KEGG)。
1.6 SSR分析
采用MISA软件对云锦杜鹃单基因簇进行简单序列重复(SSR)分析,鉴定其中6种类型的SSR;各SSR类型重复次数设定为:单核苷酸SSR,重复数≥10次;二核苷酸SSR,重复数≥6次;三至六核苷酸SSR,重复数≥5次。
2 结果与分析
2.1 测序结果与组装
分别提取无性系ZL-01组培苗根、茎和叶的RNA。检测结果表明:根RNA的质量浓度为362 mg·L-1,28S/18S为1.6,RNA完整性计数(RNA integrity number,RIN)为9.6;茎RNA的质量浓度为515 mg·L-1,28S/18S为 1.8,RIN值为 9.8;叶 RNA的质量浓度为 485 mg·L-1,28S/18S为 1.8,RIN值为9.7。这些指标均符合建库测序要求,因此,将这3个组织的RNA等量混合后进行测序。
测序数据经处理后得到94252430个干净测序片段(clean reads),包含了14.14 Gb核苷酸序列信息,GC含量为47.58%。测序质量评估结果显示,碱基错误率为0.02%,Q20为96.70%,Q30为91.85%。这些表明该转录组的数据量和质量均较高,为后续的序列组装提供了高质量的原始数据。
通过Trintiy软件组装得到112777个转录本,总长度为91621682 bp,平均长度为812.4 bp,N50为1410,其中长度在1 kb以上的有29225条,占25.92%;2 kb以上的10418条,占9.24%(表1)。对转录本进行聚类和组装得到84633个单基因簇,总长度为58517298 bp,平均长度为691.4 bp,N50为1177 bp;其中超过1 kb的有16631条,占19.65%;超过2 kb的5760条, 占6.81%(表1)。

表1 云锦杜鹃转录组组装结果Table 1 Summary of transcriptome assembly for R.fortunei
2.2 基因功能注释
将单基因簇序列与Nr,Nt,SwissProt,GO,KOG,KEGG和Pfam 7个数据库进行比对,共有35526条单基因簇获得成功注释,占单基因簇的41.97%(表2)。其中,30700条单基因簇获得Nr数据库注释,占36.27%; 23215条单基因簇获得GO数据库注释,占27.43%; 22882条单基因簇获得Pfam数据库注释,占27.03%;22202条单基因簇获得Swiss-prot数据库注释,占26.23%;18046条单基因簇获得Nt数据库注释,占21.32%;11085条单基因簇获得KOG数据库注释,占13.09%;9887条单基因簇获得KEGG数据库注释占11.68%(表2)。

表2 云锦杜鹃单基因簇功能注释Table 2 Annotation of unigenes in transcriptome of R.fortunei
在Nr库中,云锦杜鹃转录组注释到其他物种的单基因簇基因序列共30700条。其中与葡萄Vitis vinifera基因序列相似的最多,所占比例为24.39%;其次为中粒咖啡Coffea canephora,所占比例为7.39%;第3为可可Theobroma cacao,占6.77%;其他相似性序列数量大于3%的物种有烟草Nicotiana tomentosiformis(6.19%), 毛果杨Populus trichocarpa(4.09%), 甜橙Citrus sinensis(3.82%), 麻疯树Jatropha curcas(3.76%)和梅花Prunus mume(3.16%), 其他物种占 40.43%(图 1)。
2.3 单基因簇的GO功能分类
通过GO数据库的注释,共有23215条单基因簇获注释信息,得到119389个GO功能注释。由分类结果可知:生物学过程(biological process)最多55692条,占46.65%,其次是细胞组分(cellular component),35577条,占29.80%,最少的分子功能(molecular function),有28120条,占23.55%;这三大功能分类又可分为55个亚类,其中生物学过程23个亚类,细胞组分18个亚类,分子功能14个亚类(图2)。生物学过程中,涉及细胞过程、代谢过程和单一有机体进程的单基因簇较多,分别有12698,12376和9581条;细胞组分中涉及较多的是细胞、细胞部分和大分子复合体,分别有7216,7214和4713条;分子功能中涉及较多的有结合功能和催化活性,分别有13302,10524条(图2)。

图1 云锦杜鹃单基因簇Nr数据库比对相似性物种分布图Figure 1 Similarity of unigenes of R.fortunei with those of other species in Nr database

图2 云锦杜鹃单基因簇的GO功能分类Figure 2 GO functional categories of R.fortunei unigenes
2.4 单基因簇的KOG功能注释
通过KOG数据库对云锦杜鹃单基因簇进行注释,结果显示有11085条序列获得12475个注释信息,可分为26个功能分类(图3)。从基因功能分类来看,涉及一般功能预测的序列最多,多达1970条;涉及翻译后修饰、蛋白翻转、分子伴侣功能的序列次之,有1527条;而涉及核结构、胞外结构和细胞运动的序列很少,仅有37,34和5条(图3)。

图3 云锦杜鹃单基因簇的KOG功能分类Figure 3 KOG functional categories of R.fortunei unigenes
2.5 KEGG通路注释
利用KEGG注释系统对云锦杜鹃单基因簇涉及的代谢途径进行分析,结果显示:9887条单基因簇得到15455个注释,归属于272条通路。按获得注释的基因数量进行排序,取前20个途径,发现含有200条单基因簇以上的通路有10个,涉及碳代谢的单基因簇最多,有377条;其次是与核糖体相关的单基因簇,有364条;第3位是与氨基酸合成相关的单基因簇,有348条;涉及其他途径有内质网蛋白加工(319)、剪切体(256)、植物激素信号转导(244)、淀粉和蔗糖代谢(240)、RNA转运(227)、氧化磷酸化(224)和植物病原物相互作用(210);其他通路的单基因簇数量均在200以下(表3)。

表3 云锦杜鹃单基因簇的KEGG分析Table 3 Summary KEGG pathway of R.fortunei transcriptome
2.6 MADS-box基因家族分析
MADS-box基因家族在植物花分生组织形成、花器官发育等过程中发挥关键作用[16]。鉴定转录组中的MADS-box基因有助于进一步研究或调控云锦杜鹃成花过程。通过与Nr,Nt和Swiss-prot三大数据库比对,共找出编码MADS-box基因的单基因簇序列24条,分别属于10个不同的亚家族(表4)。其中AGL17亚家族成员最多,包含c41091_g4,c39737_g1,c36841_g1,c38538_g1和c40334_g4;SQUA和SVP亚家族则各有 4个成员,分别为 c50592_g1,c10093_g1,c9572_g1,c2583_g1与 c37773_g1,c31351_g1,c30064_g1,c33061_g2;TM3亚家族有3个成员,其他亚家族仅有2个或1个成员(表4)。这些单基因簇的同源基因分别与花分生组织发育、花期调控、花器官发育、果实发育等重要生物学过程相关。
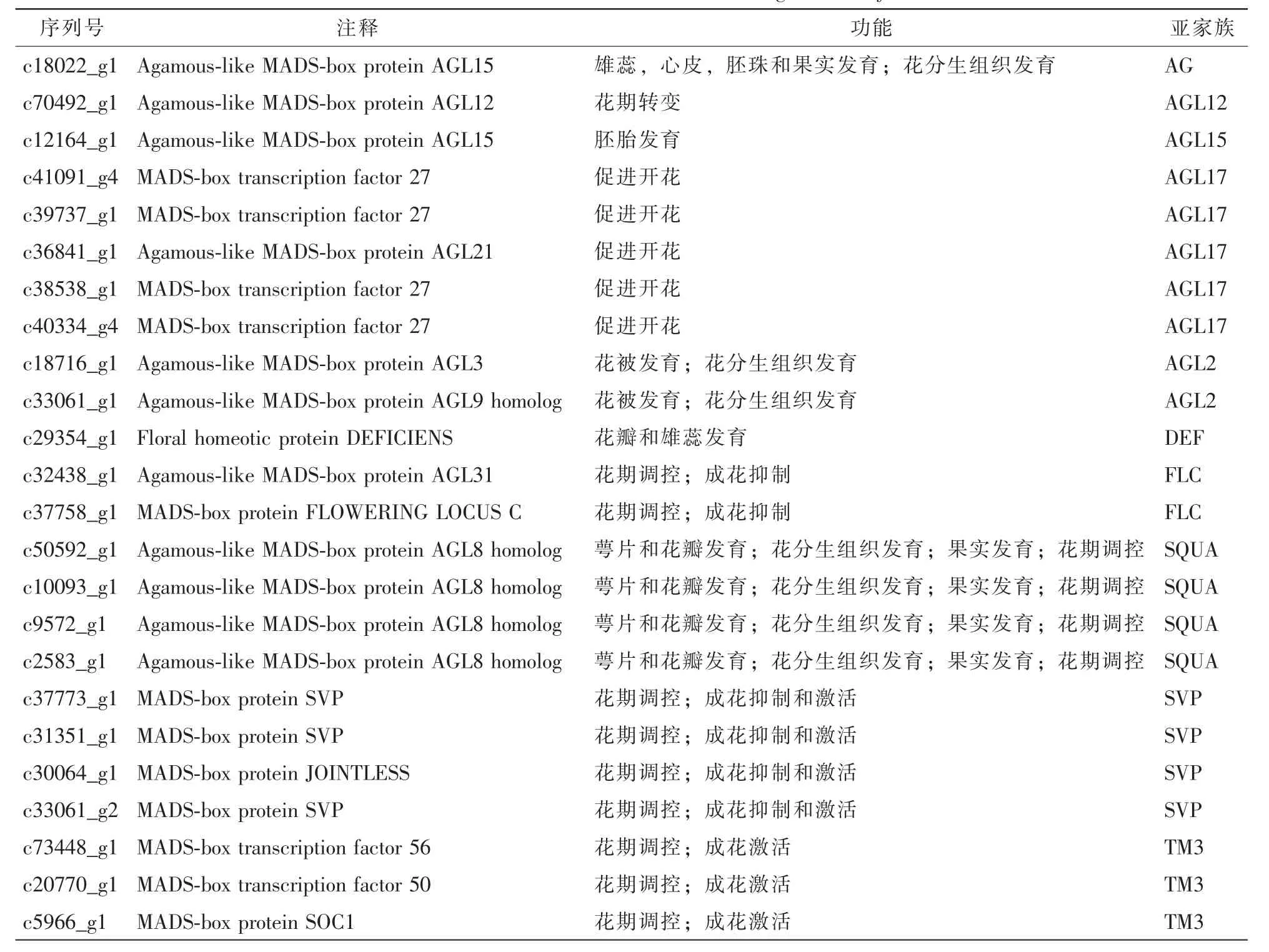
表4 云锦杜鹃成花相关MADS-box基因鉴定Table 4 Identification of Floral related MADS-box genes of R.fortunei
2.7 SSR分析
利用MISA软件对云锦杜鹃转录组序列进行分析,共发现21900个SSR位点,分布在17414条单基因簇中,其中有含有1个以上SSR位点的单基因簇有3606条(表5)。在所有SSR位点中,双碱基重复SSR最多,有12294个,占总数的56.14%;其次为单碱基重复SSR,有6448个,占总数的29.44%;三碱基重复SSR有2970个,占13.56%;四碱基重复SSR有140个,占0.63%;五碱基和六碱基重复SSR分别仅有25个和23个(表5)。进一步分析这些SSR重复基序,可以发现,在单碱基SSR中,A/T发生频率最高;双碱基SSR中,发生频率最高的是AG/CT,其次是AC/GT;三碱基重复中发生频率最高是AAG/CTT, 其次是AGG/CCT(图4)。
3 结论与讨论
中国是世界上杜鹃花属植物资源最为丰富的国家,为世界杜鹃花育种做出了巨大贡献。但中国杜鹃花育种尤其常绿杜鹃育种水平较欧美、日本等国仍有较大差距,资源开发利用水平低,优良品种少。云锦杜鹃作为中国特有常绿杜鹃,观赏价值高、抗性好,野生资源也较为丰富,具有良好的开发潜力。但由于缺乏相关的遗传背景信息,云锦杜鹃的遗传多样性、杂交子代鉴定和优异基因型挖掘等遗传育种研究一直受到制约。近年来,随着高通量测序技术的发展,转录组测序已成为非模式生物遗传背景解析的重要手段,在标记开发、表达分析、功能基因挖掘等方面得到广泛应用[17-21]。本研究利用Illumina测序技术对云锦杜鹃组培苗的转录组进行测序和分析,以获得其转录组序列信息。云锦杜鹃转录组的测序数据分析结果表明:数据的Q30值为91.85%,拼接后共获得84633条单基因簇,平均长度为691.4 bp,N50值为1177 bp。一般认为Q30在80%以上就认为测序质量可靠;N50值越大就表示长片段越多,且不小于800 bp就说明组装得到序列完整性较好[22]。上述结果表明本研究测序数据的质量和组装长度达到了转录组分析的基本要求,为进一步分析利用奠定了基础。

表5 云锦杜鹃转录组SSR分析结果Table 5 Summary of SSR in R.fortunei transcriptome
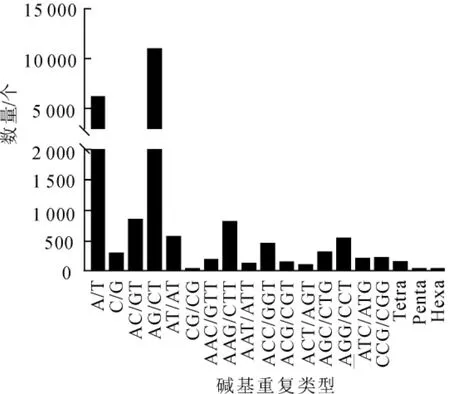
图4 云锦杜鹃SSR类型数量分布Figure 4 Distribution of SSR motif number of R.fortunei
基因功能注释是转录组分析的重要内容,是进行重要功能基因挖掘的前提。因此,本研究利用Nr和Swiss-prot等七大数据库对云锦杜鹃转录组序列进行功能注释,结果表明:共有35526条单基因簇获得注释信息,仍有约5万条序列没有获得注释。这与薏苡Coix lachryma-jobi[20]和岩穴蕨Monachosorum maximowiczii[23]的情况类似,可能是由于云锦杜鹃是未测序物种,在相关数据库中缺乏对应的功能注释信息,也可能是部分云锦杜鹃单基因簇序列本身太短造成的。GO和KOG注释功能分类的结果显示,云锦杜鹃单基因簇的功能涉及了各类生命活动;KEGG通路注释到9887条单基因簇,涉及到272条代谢通路。这些结果表明:对于云锦杜鹃等这一非模式植物,转录组测序可以有效地解析遗传背景,获得大量序列信息。基因功能注释也是挖掘与特定途径或功能相关基因的有效手段。如在紫色黄秋葵Abelmoschus esculentus中,通过转录组的KEGG注释,获得与花色素苷、黄酮、类黄酮、二萜类和萜类骨架等生物合成相关的单基因簇[24]。本研究通过功能注释,也鉴定获得24个编码MADS-box基因的单基因簇,属于10个不同的亚家族,它们可能与花分生组织发育、花期调控、花器官发育等重要成花过程相关。
简单序列重复(SSR)又称微卫星序列,具有共显性、密度大、信息量丰富等优势,广泛应用于遗传图谱构建、遗传多样性分析、基因定位、分子标记辅助育种等方面[25]。利用转录组序列开发SSR标记具有通量高,成本低的优势,已在多种植物中获得成功[19,26-27]。在大王杜鹃R.rex转录组序列中鉴定获15314个SSR位点,占比最高的为双碱基重复SSR,其次为单碱基重复SSR和三碱基重复SSR,且利用这些SSR位点开发了相应引物对20份大王杜鹃种质进行了遗传多样性评价[3]。本研究也在云锦杜鹃单基因簇序列中鉴定获得21900个SSR位点,发现其中双核苷酸重复SSR最多,达到12294个;其次为单核苷酸重复和三核苷酸重复SSR,这与大王杜鹃中发现的规律类似。这些结果将为云锦杜鹃SSR标记开发提供重要序列信息,也为云锦杜鹃种质资源遗传多样性分析、功能基因挖掘以及分子辅助育种等工作提供了重要基础。
猜你喜欢
华人时刊(2022年15期)2022-10-27
中国计量大学学报(2022年2期)2022-07-18
山东第一医科大学(山东省医学科学院)学报(2022年1期)2022-02-28
健康之家(2021年19期)2021-05-23
科学之谜(2021年2期)2021-04-25
科技与创新(2020年12期)2020-11-28
教学考试(高考生物)(2020年6期)2020-11-23
华人时刊(2020年13期)2020-09-25
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
