
SrCoO3的电子结构、弹性及热力学性质的第一性原理计算
2019-11-26 08:55李文涛
陕西科技大学学报 2019年6期
李文涛
(陕西科技大学 文理学院, 陕西 西安 710021)
0 引言
含有过渡金属元素的钙钛矿氧化物,由于其结构中电子的自旋、轨道和晶格等自由度相互关联和竞争,可以表现出丰富的物理性质,如二维自由电子气、蓝光发射、超导、铁磁、铁电和多铁等物理性质[1],在传感、信息存储和电子自旋器件等领域拥有广阔的应用前景.含Co的钙钛矿氧化物及其衍生结构如LaCoO3和SrCoO3等,由于Co元素中3d电子可能存在多种自旋态,并且同材料的晶相、应变、掺杂以及缺陷等密切相关,近年来也一直受到广泛关注[2,3].此外,有研究报道发现这类氧化物,如SrCoO3-δ、La0.6Sr0.4CoO3和SrCo0.9Sb0.1O3-δ等,还可作为中低温固态燃料电池(SOFC)的电极材料,表现出较好的性能和应用前景[3,4].

鉴于SrCoO3基态的电子结构和铁磁序等还存在一定的争议;同时,关于其弹性、声子以及热力学性质等的研究还有明显不足.因此,本文针对立方相的SrCoO3,利用第一性原理计算方法,阐明其晶格常数和电子结构,分析其铁磁性质,并给出了弹性、声子和热力学性质的理论计算结果,从而为今后针对这类材料的实验研究和应用提供理论依据.
1 计算方法
1.1 电子结构和弹性性质
本文的第一性原理计算是基于密度泛函理论,通过平面波赝势方法的Quantum-ESPRESSO(QE) 程序包实现的[8].为了准确描述Co原子中3d电子间的关联作用,计算中采用广义梯度近似下的GGA+U的方法,其中U为描述Hubbard Coulomb相互作用的参数,根据文献结果将U取为6 eV[5].晶胞中价电子与离子核间的相互作用通过超软赝势(USPP)来描述.为了分析铁磁性质,计算过程中考虑了电荷密度的自旋极化.程序中所有参数在使用前都经过了收敛测试,此时平面波的截断能为816 eV,布里渊区内k点采用8×8×8的Monkhorst-Pack网格.自洽计算的收敛精度为总能量的变化低于10-8eV,晶胞内各原子的受力均小于0.01 eV/Å.
弹性计算是基于Lagrangian应变理论,结合晶胞空间群类型,使用QE程序计算晶胞在一系列应变后体系的能量值,再利用能量对Lagrangian应变的二阶导数计算得到弹性常数[9].利用弹性常数就可以进一步计算得到材料的体弹性模量、剪切模量、杨氏模量和泊松系数[9].
1.2 声子和热力学性质
声子谱的计算是基于密度泛函微扰理论DFPT方法实现的(QE程序中ph.x及相关模块).计算过程中,布里渊区内格波的波矢q采用6×6×6相对Γ点无偏移网格,电子波函数的k点采用8×8×8的Monkhorst-Pack网格.
热力学性质的计算采用晶格振动谐振近似下的德拜模型,分别使用了Debye-Slater和Debye-Grüneisen模型,后者考虑了材料泊松系数随体积的变化[10].
2 结果与讨论
2.1 晶格常数与弹性性质
立方相的SrCoO3为典型的钙钛矿结构,其晶胞结构示意图如图1中插图所示.Co原子占据体心位置,面心位置处的O原子和体心的Co原子形成CoO6八面体结构,Sr原子则占据立方体的顶点位置.
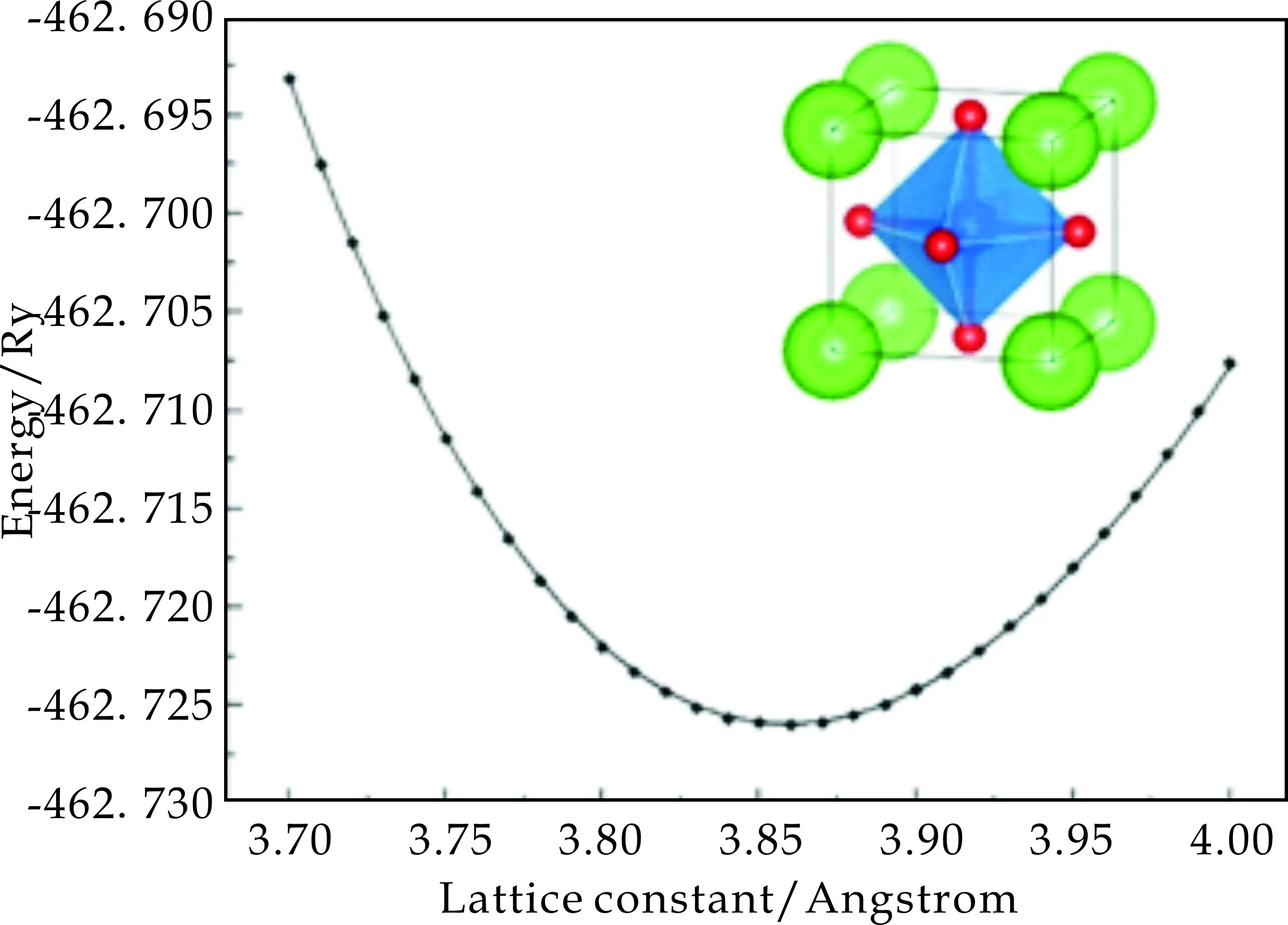
图1 立方相SrCoO3的能量随晶格常数的变化
为了得到立方相SrCoO3的晶格常数,首先计算了一系列离散晶格常数下晶胞的能量值,并利用这些计算结果进行物态方程的拟合.图1给出了立方相SrCoO3体系能量随晶格常数变化的理论计算值(图中离散点)和拟合结果.其中拟合曲线是根据能量随体积变化的4阶Birch-Murnaghan物态方程实现的[10].通过计算结果的拟合就可以得到温度和压强为0时立方相SrCoO3的晶格常数和体弹性模量.计算结果如表1所示,其中a、B和Bp分别为计算得到的晶格常数、体弹性模量和体弹性模量对压强的导数.表1中也分别给出了GGA和GGA+U方法的计算结果.

表1 立方相SrCoO3的晶格常数与体弹性模量
文献报道中关于SrCoO3立方相的晶格常数以及体弹性模量也在表格中给出[11,12].结合表1的数据,发现无论晶格常数还是体弹性模量,使用GGA+U方法计算得到的结果更加接近于实验测量值,而仅用GGA方法得到的晶格常数则偏小.
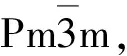

表2 立方相SrCoO3的弹性性质(单位:GPa)
从计算结果可以看出,利用弹性常数计算得到的体弹性模量B'和表1中利用物态方程拟合得到的B非常接近,并和表1中给出的文献报道值也比较一致,这也进一步验证了弹性常数计算的可靠性.目前文献报道中关于SrCoO3立方结构的二阶弹性常数Cij的实验数据还不够完备,但是也有利用改进后的刚性离子模型(modified rigid ion model)计算得到的数值结果[12,14].其给出的体弹性模量值(132 GPa)和本文结果比较一致,但是在C11和C12数值的计算结果上却存在一定的差异[12].这可能是由于前者使用的计算方法中存在较多假设和经验参数的设置,同时,本文中的第一性原理计算方法也并未考虑温度和压强等对SrCoO3弹性常数的影响.
2.2 电子结构
立方相SrCoO3的能带结构如图2所示.图中所有能量以费米能级作为参考原点,并用虚线标出了费米能级的位置.由于在计算过程中考虑了电荷密度的自旋极化,所以在图2中分别用黑色实线和红色虚线给出了两种自旋方向的能带结构.从图2中可以清晰地看出,两种自旋的能带结构拥有较大的不同.特别是在费米能级附近,能带中自旋向下的部分和自旋向上的部分相比较,色散特征比较一致,但是有能量向上平移的特征.这说明在费米能级附近,电子更倾向于占据自旋向上的电子态.这也揭示了立方相SrCoO3的铁磁性质,而自旋向上是其铁磁极化方向.根据理论计算结果,晶胞中每个Co原子的平均磁矩为3.13μB,这也与SrCoO3的中间自旋态(理论自旋态S=3/2)比较一致.
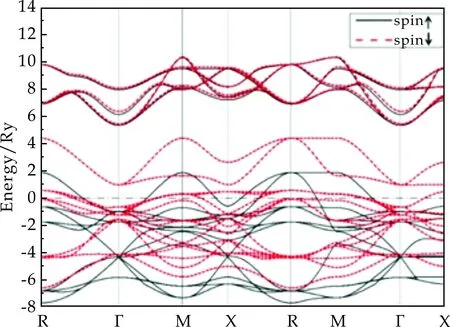
图2 SrCoO3立方相的能带结构
从图2还可看出,无论是哪种自旋方向,都有能带穿过费米能级.这说明两种自旋都存在未被电子完全占据的能带结构,因此皆表现为金属性.这与之前文献报道中关于SrCoO3是铁磁半金属的性质不太一致[6],但是从图2中也可看出,对于主自旋方向的电子,在费米能级上方存在一个带隙,这表明通过掺杂、门电压调控等手段来调节费米能级的位置,就可能使SrCoO3成为铁磁半金属,此时,SrCoO3的自旋态也会发生改变.
为了揭示晶胞中各原子轨道对费米能级附近电子态的贡献,图3给出了立方相SrCoO3不同自旋方向的电子态密度,包括总态密度和各轨道的部分态密度,其中所有能量皆以费米能级作为参考原点.
从图3可以看出,两种自旋的态密度在费米能级附近有很大的不同,这和图2中能带计算的结果相一致.对于自旋向下的电子,即图3的下半部分,费米能级附近态密度主要来源于Co的3d电子的贡献,并且其态密度有峰值出现在费米能级上方,说明电子并未完全占据;对于自旋向上的电子,即铁磁极化方向,Co的3d轨道主要在费米能级下方,说明大部分被电子占据,这也揭示了SrCoO3的自旋极化主要来源于Co原子3d电子的不同填充.
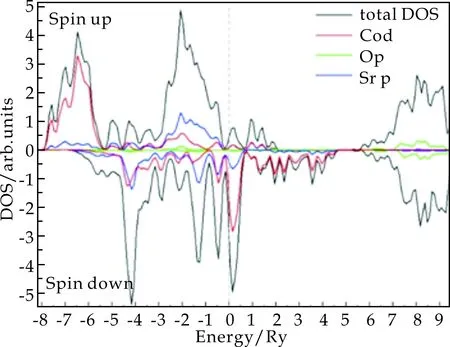
图3 SrCoO3立方相的电子态密度
由于钙钛矿结构中体心的Co原子和面心处的O原子形成的CoO6八面体结构,因此根据晶体场理论,Co原子的3d轨道会劈裂形成3个t2g轨道和2个eg轨道[5].因为SrCoO3中Co为+4价,所以3d轨道上有5个电子.当SrCoO3处于中间自旋态时,3d电子的占据态为t2g4eg1:自旋向上的电子占据了3个t2g轨道和1个eg轨道;自旋向下的电子占据了1个t2g轨道.图4给出了两种自旋的t2g轨道和eg轨道的部分态密度.

图4 SrCoO3中Co原子t2g和eg轨道部分态密度
从图4可以看出,对于自旋向上的电子,t2g轨道被完全占据,eg轨道则穿过费米能级;对自旋向下的电子,t2g轨道和eg轨道大部分在费米能级以上.这与SrCoO3中3d电子处于中间自旋态相一致,也与SrCoO3晶胞磁矩为3.13μB的计算结果吻合.
2.3 声子计算

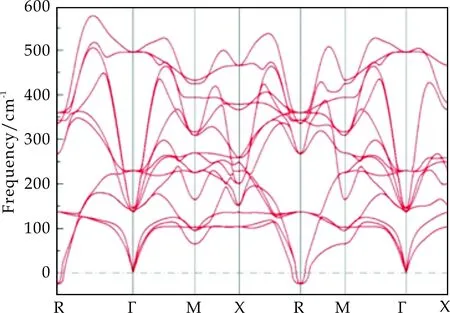
图5 立方相SrCoO3晶格振动声子谱
从图5可以看出,立方相SrCoO3在Γ点处不存在软膜,但是在R点存在软膜,这与SrTiO3声子谱在R点出现的软膜相类似[16].由于SrTiO3在低温下会发生立方相到四方结构的相变,而R点软膜的出现意味着低温下四方相的出现[17].因此,图5声子谱的计算结果表明SrCoO3的立方相在低温下可能并不是稳定结构,这与之前文献报道中关于四方结构的SrCoO3晶格在低温下更为稳定的结论一致[5].
2.4 热力学性质
由于立方相SrCoO3的声子谱在R点存在少量的虚频,因此就不再适合利用声子谱态密度的准谐振近似方法计算其热力学性质.这里采用晶格振动谐振近似下的德拜模型,分别利用Debye-Slater和Debye-Grüneisen模型计算SrCoO3不同温度和压强下的热力学性质.其中Debye-Grüneisen模型考虑了泊松系数随体积的变化,而Debye-Slater模型在计算中使用固定的泊松系数[9].根据之前SrCoO3弹性性质的计算结果,Debye-Slater模型中将SrCoO3的泊松系数取值为0.36.由于在热力学性质的计算过程中使用了两种不同的德拜模型,所以在图6到图9中分别使用实线和虚线来表示Debye-Grüneisen模型和Debye-Slater模型的计算结果.立方相SrCoO3的晶格常数和体弹性模量随温度和压强的变化分别如图6和图7所示.

图6 SrCoO3不同温度和压强下的晶格常数
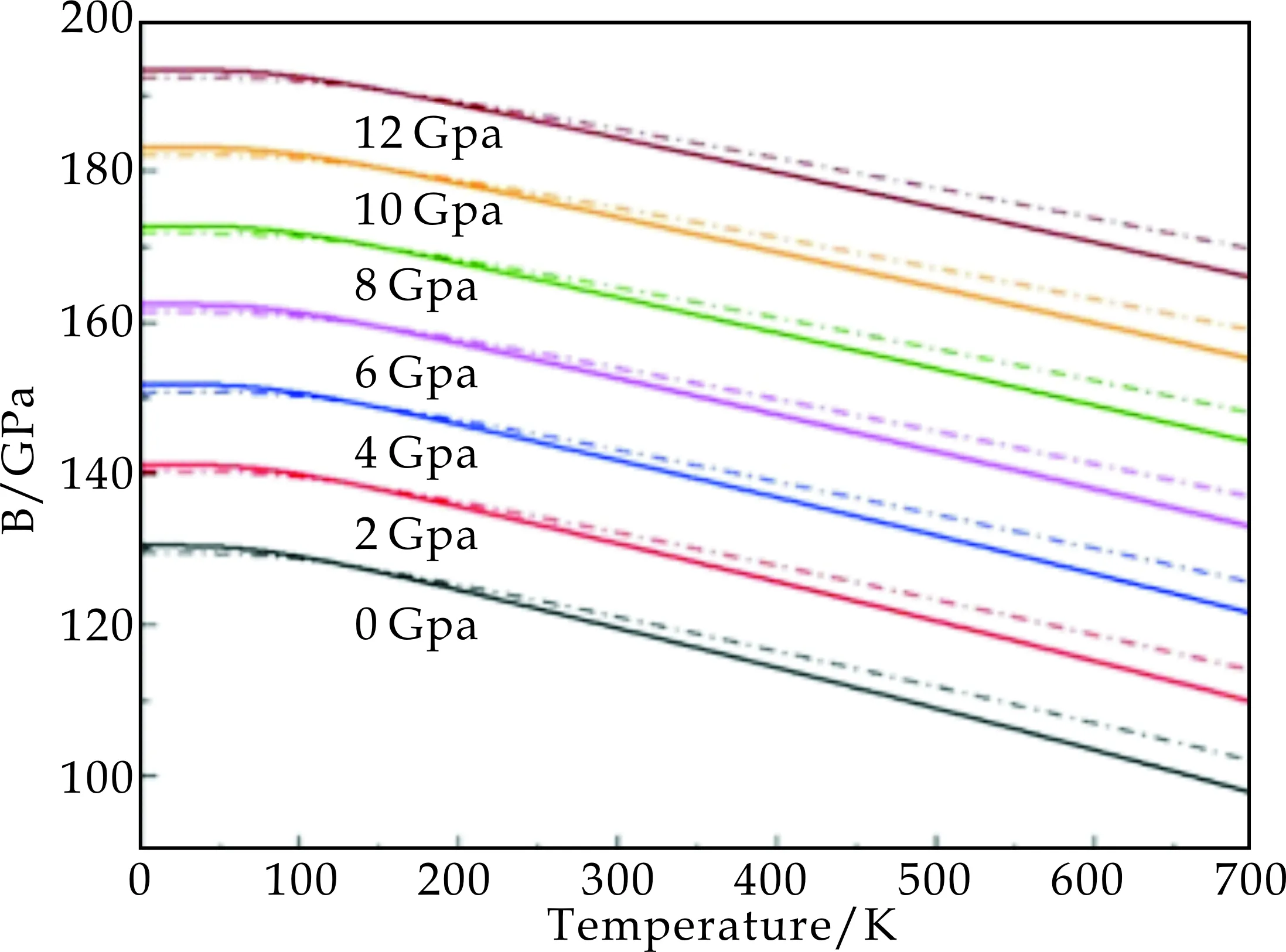
图7 SrCoO3不同温度和压强下的体弹性模量
图6和图7中温度变化的范围为0 K~700 K,并用不同颜色的曲线给出了压强从0 GPa~12 GPa的计算结果.两种德拜模型都反映出SrCoO3的晶格常数随温度升高而增大,并且随压强增大而降低的趋势;体弹性模量则随着温度升高而降低,随压强升高而增大,正如图7中所示.通过对比两种模型的计算结果,可以发现在低温区间两种模型的差别较小,而当温度大于200 K时差别增大.这也说明随着温度升高,泊松系数随体积的变化需要在德拜模型中考虑.
立方相SrCoO3的比热容随温度和压强的变化如图8所示.图8不仅包括了定压比热容Cp的计算结果,其中的插图还给出了定容比热容Cv随温度和压强的变化.可以看出两种比热容在低温下均趋近于0,随着温度升高,Cv则趋近于热力学Dulong-petit极限,如图8内插图中虚线所示.图9给出了立方相SrCoO3的热膨胀系数随温度和压强的变化.从图8中可以看出,相同温度下,随着压强的增大,热膨胀系数则变小.当温度低于200 K时,热膨胀系数随温度的升高有快速的增长,但是随着温度继续升高,则增长速度开始变缓.根据文献中LaCoO3的计算结果[18],这种温度变化规律并不是由于德拜模型的近似造成的.

图8 SrCoO3不同温度和压强下的比热容
图8和图9的计算结果还表明,无论是比热容还是热膨胀系数,在相同的温度和压强下,采用Debye-Grüneisen模型得到的计算结果比用Debye-Slater模型的结果数值偏大,特别是对热膨胀系数,两者的计算结果存在较大的差别.

图9 SrCoO3不同温度和压强下的热膨胀系数
3 结论
本文针对SrCoO3立方相的电子结构、弹性以及和晶格振动有关的声子和热力学性质等进行了第一性原理计算研究.通过计算给出了立方相SrCoO3的晶格常数、弹性常数以及体弹性模量、剪切模量等参数.通过对自旋极化后电子的能带结构和态密度的分析,研究了SrCoO3的铁磁性质,计算结果表明SrCoO3为铁磁金属,晶胞内平均磁矩为3.13μB,并且Co原子的3d电子处于中间自旋态.通过晶格振动声子谱的计算,发现立方相的SrCoO3在布里渊区R点处存在软膜.结合文献中关于SrTiO3声子谱的讨论,揭示了SrCoO3的立方相在低温下并非稳态结构,可能存在四方结构的相变.最后,采用晶格振动谐振近似下的德拜模型,分别利用Debye-Grüneisen和Debye-Slater两种模型计算了立方相SrCoO3不同温度和压强下的热力学性质,发现当温度大于200 K后,两种模型的计算结果有较大差异.这说明当温度较高时应该采用更为准确的Debye-Grüneisen模型.
猜你喜欢
数学物理学报(2022年5期)2022-10-09
太原理工大学学报(2022年5期)2022-09-23
当代作家(2021年11期)2021-12-17
中外文摘(2021年7期)2021-04-23
科学(2020年4期)2020-11-26
江苏科技大学学报(自然科学版)(2020年5期)2020-11-17
发明与创新·小学生(2020年10期)2020-10-19
科学(2020年4期)2020-01-11
发明与创新·小学生(2019年12期)2019-12-05
学生天地·小学低年级版(2016年9期)2016-05-14
