
理论研究4′-甲氧基-3-羟基黄酮的激发态分子内质子转移机制
2019-11-29 08:12徐昊浩刘松松宋玉志
山东师范大学学报(自然科学版) 2019年4期
徐昊浩 刘松松 张 慧 周 勇 宋玉志
( 山东师范大学物理与电子科学学院,250358,济南 )
1 引 言
氢键是一种既可以形成于分子间又可以形成于分子内的弱静电相互作用[1].它可以被表达为X-H...Y的形式,其中X是氢供体,Y是氢受体,像氮,氧,氯和氟等一些带负电的原子都可以作为X和Y.研究表明自然界的很多物质中都存在氢键,如DNA,水,醇类,蛋白质等.因此氢键已经成为各项基础研究的关键媒介[2-4].最近,韩等人提出了激发态氢键增强的机制[5],这为激发态分子内质子转移的研究提供了理论支持.质子转移通常以氢键为桥梁,发生在具有酸性或者碱性基团的分子中.ESIPT分子中的质子给体和受体基团距离很近,受到激发后在强氢键的作用下电荷会重新排布,同时氢原子由给体基团转移到受体基团从而完成ESIPT过程[6].激发态质子转移过程是在1956年Weller及其同事进行水杨酸实验时首次观察到的[7-9],之后便引起了研究激发态分子内和分子间质子转移的热潮.与普通荧光团相比,ESIPT发色团最显著的光物理特性是大的斯托克斯位移,这可以避免自吸收和内部滤光器效应[10].同时,ESIPT是一种亚皮秒级的超快反应过程,烯醇到酮的转变将经历一个四级光循环系统[11].由于烯醇和酮的相互转变,分子的物理和化学性质也会发生巨大的变化,尤其体现在光谱行为中. 这类分子的荧光光谱一般会出现两个峰:分子未发生质子转移之前的发射峰和分子发生质子转移之后的发射峰.一般来说由于分子发生质子转移后的光谱行为相对于质子转移前会产生显著的红移现象,所以荧光光谱中波长较长的峰为分子发生质子转移之后的荧光发射峰.基于这种特性,ESIPT分子被广泛应用于荧光探针、荧光遥感技术、分子开关和激光染料等领域[12-16].
基于ESIPT特性的3-羟基黄酮衍生物由于其荧光发射对环境极性和电场的出色灵敏度使得科研工作者对其产生了浓厚的研究兴趣.Skilitsi等人最近设计合成了4'-甲氧基-3-羟基黄酮, 与3-羟基黄酮相比,其在2-苯基部分的对位带有甲氧基.该化合物最近被官能化为合成氨基酸,为蛋白质相互作用研究提供了非常有前途的荧光特性[17].同时,Sholokh等人最近的工作证明了上述化合物在开发用于检测生物分子相互作用的高效荧光探针方面的成功[18].虽然实验上得到了4M3HF的光谱特性,但并没有在理论上定量地解释其ESIPT机理,因此我们致力于去研究4M3HF的氢键动力学过程以及ESIPT机制.我们的工作采用了密度泛函理论(DFT)和含时密度泛函理论(TDDFT)方法,获得的前线分子轨道是被用来表征电子密度的变化,计算的几何参数,红外振动光谱和降低密度梯度将用于讨论分子内氢键的特征.此外,基态和第一激发态的势能曲线也通过柔性扫描获得.
2 理论方法
全部的理论计算都是在Gaussian 16软件中[19]基于DFT和TDDFT方法进行的[20].同时,我们采用B3LYP泛函和6-31g(d)基组来进行分子的理论模拟.为了与实验中的溶剂环境一致,我们将4M3HF置于IEF-PCM溶剂化模型中的甲醇溶剂中来进行结构优化,用以得到最稳定的分子几何构型,并且不对键长、键角和二面角进行限制.频率分析表明没有虚频存在,因此优化得到的分子构型全部处于最稳定的状态.为了定量的给出分子的反应路径,我们以O1-H2之间的距离为变量构建了分子基态与激发态的势能面,其中以每步0.03 Å的距离从0.9 Å扫描到了2.15 Å.
3 计算结果与分析
3.1分子几何构型的优化我们在甲醇溶液中分别优化了在基态(S0)和第一激发态(S1)中4M3HF的几何构型,获得的两种同分异构体如图1所示,分别是烯醇(enol) (a)和酮式结构(keto) (b).为了更加清楚的描述接下来的分析,我们将与氢键相关的原子进行了标序.计算得到的键参数被陈列于表格1中.对于烯醇式结构,O1-H2的键长在基态下为0.98 Å,在第一激发态增长到了1.01Å,在光的激发下H2-O3的键长从1.95 Å缩短到了1.78 Å,同时O1-H2…O3的键角从121.1°增加到126.7°.键长的变化以及键角的增加充分说明了分子内氢键O1-H2…O3在第一激发态得到了加强.对于酮式结构,O1-H2的键长从第一激发态到基态由1.98 Å减少到1.82 Å,而H2-O3键长由0.99 Å伸长为1.01 Å,O1…H2-O3的键角也从119.4°增加到125.2°.经过比较可以说明酮式结构在基态更容易发生反质子转移回到最初的烯醇式构型.
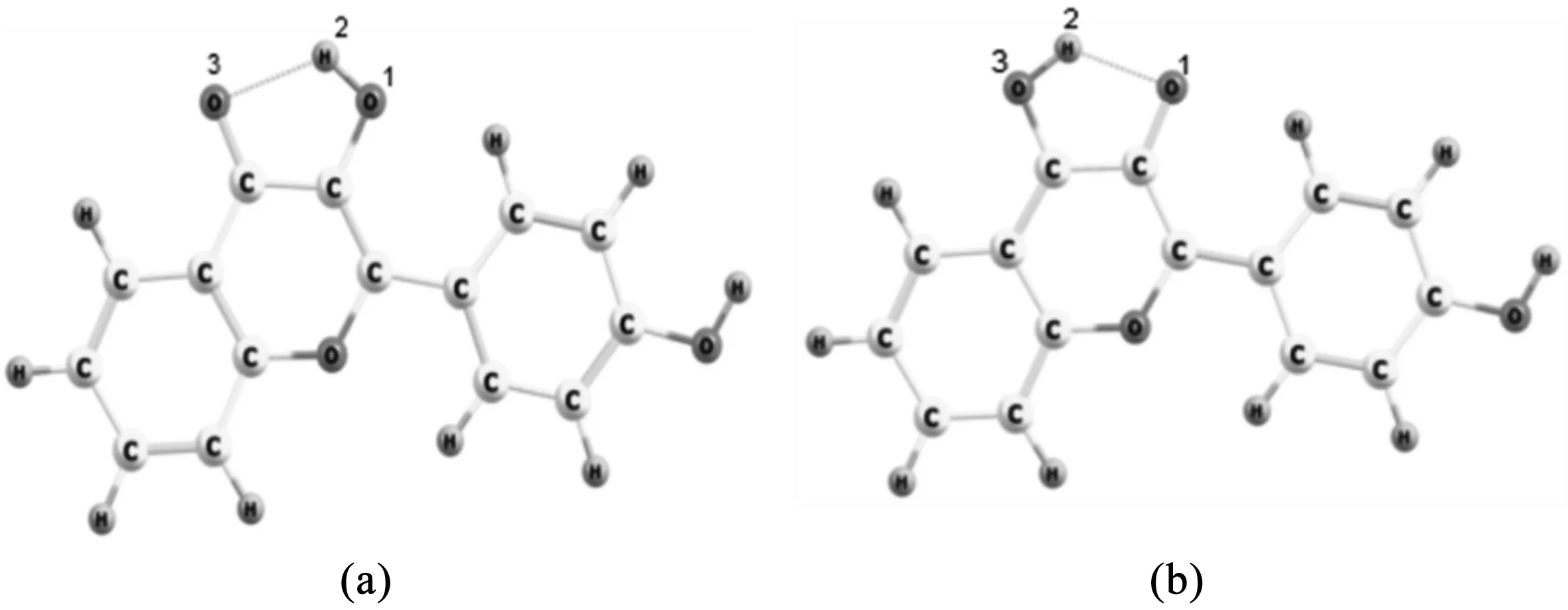
图1 优化得到的4M3HF的烯醇式结构(a)和酮式结构(b)
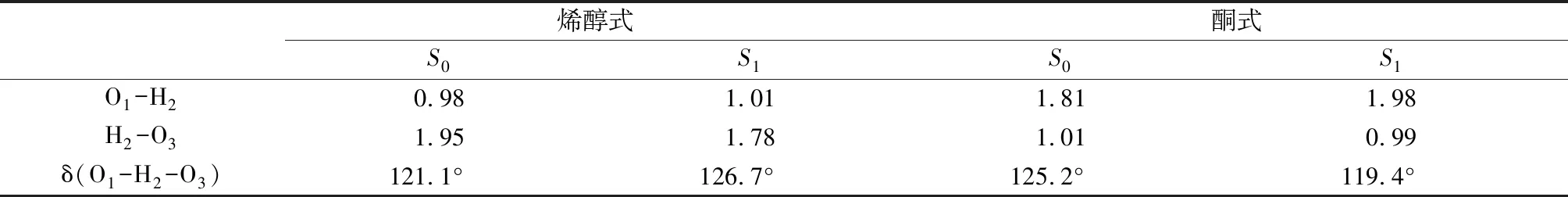
烯醇式酮式S0S1S0S1O1-H20.981.011.811.98H2-O31.951.781.010.99δ(O1-H2-O3)121.1°126.7°125.2°119.4°
此外,研究与氢键有关的化学键的红外振动光谱也是一种观察氢键强度变化的有效的方法,可以通过峰值的改变来说明氢键在基态与激发态之间发生的变化.振动频率变小(即发生红移),则氢键增强,反之则减弱.基于上述理论计算获得的4M3HF的烯醇式结构在基态和第一激发态的红外振动光谱如图2所示.在基态O1-H2的伸缩振动频率位于3 472 cm-1处,而在第一激发态O1-H2的伸缩振动频率变化到了3 077 cm-1处.红外振动光谱图上显示出的395 cm-1的红移说明分子内氢键O1-H2…O3在第一激发态有了明显的加强.
3.2前线分子轨道和电子光谱为了更好的说明4M3HF的电荷分布与转移特性,我们进行了前线分子轨道(最高已占据分子轨道(HOMO)和最低未占据分子轨道(LUMO))的相关计算与分析.烯醇式分子的HOMO和LUMO的跃迁轨道图如图3所示.分子低单重激发态的电子激发能及相应的振子强度和跃迁轨道贡献由表2给出.对于第一激发态来说振子强度最大为0.571 0,且涉及到这两个轨道之间的跃迁贡献率为97.8%.从图中可以看出HOMO轨道是π型轨道,而LUMO轨道是π*型轨道.也就是说,第一激发态具有ππ*跃迁特征.此外,该分子的电荷在HOMO和LUMO上的密度分布是不同的,这说明了第一激发态是分子内电荷转移态.从HOMO跃迁到LUMO之后,羟基部分电荷分布减少,相邻氧原子电荷分布有了明显的增加.因此,与基态相比,羟基上的质子变得酸性更强,同时羰基氧也具有更强的碱性.羰基氧对质子的吸引力势必会增加,分子间氢键O1-H2…O3也便同时增强.毫无疑问,这是质子转移的一个非常重要的积极因素.此外,为了证实计算方法的可靠性,基于优化的基态构型模拟的4M3HF的电子光谱是被绘制在图4中.计算的从基态到第一激发态的吸收峰为365 nm,这与实验文章给出的吸收峰位于350~355 nm处是相吻合的,与实验现象相同的是,模拟计算也得到了两个荧光发射峰,分别是位于432 nm处的醇式结构的发射峰和位于576 nm处的酮式结构的发射峰,他们同样与实验获得的两个位于428 nm与530 nm的荧光发射峰符合得很好.
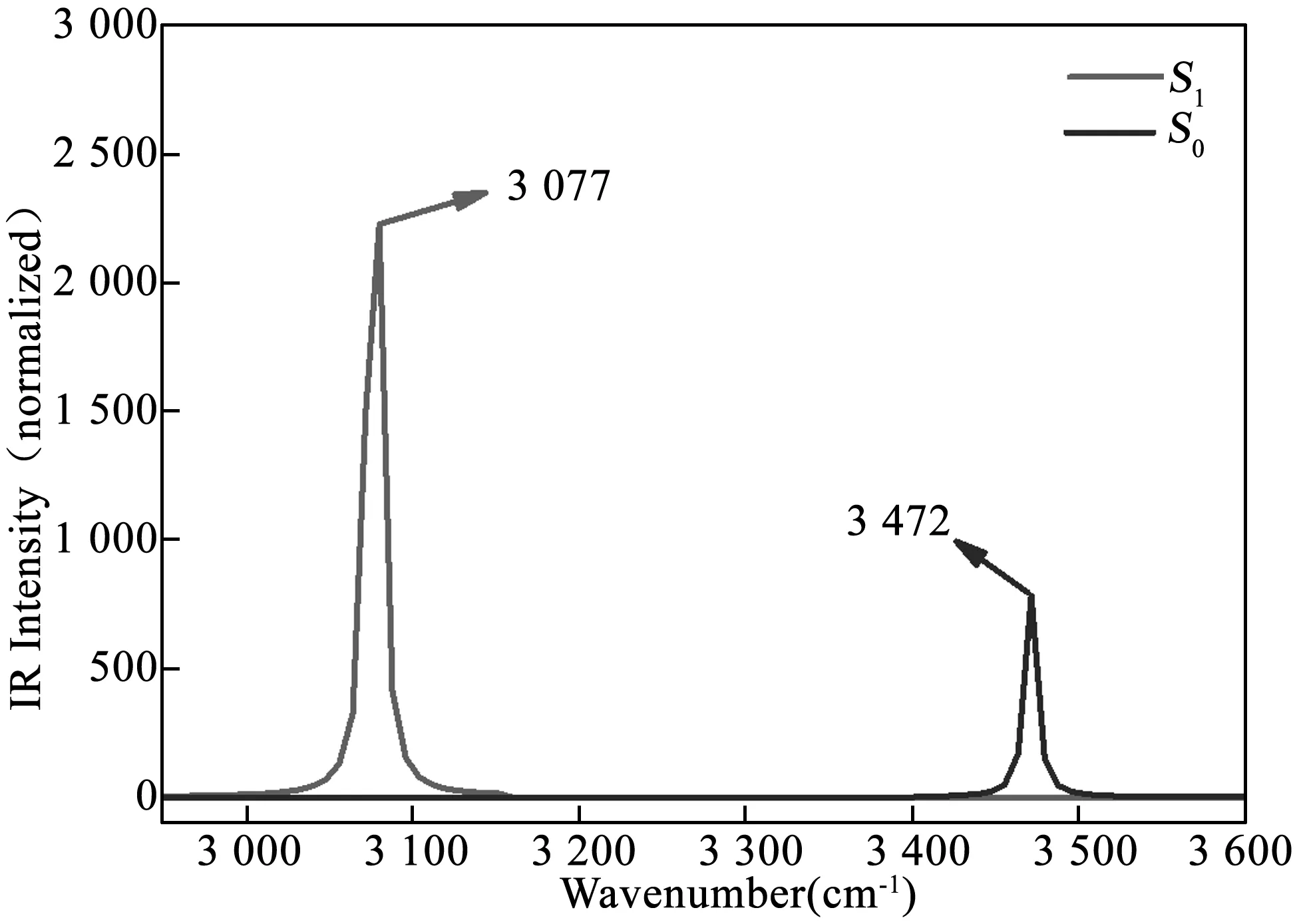
图2 4M3HF的烯醇式结构中O1-H2键在基态与第一激发态的红外振动光谱

图3 4M3HF的前线分子轨道

图4 4M3HF的吸收和发射光谱

Transitionλ(nm)ƒCompositionCI(%)S0→S13650.571 0H→L97.8S0→S23020.093 4H-3→L97.3S0→S32980.000 0H-1→L51.9
3.3约化密度梯度函数(RDG)RDG分析方法不仅可以指出哪里存在弱相互作用,还可以可视化地了解弱相互作用的强度与类型.其中,ρ(r)是总的电子密度,它只可以反应弱相互作用的强度,弱相互作用的类型需要Sign(λ)来表示.根据分子中的原子理论,RDG函数以及电子密度矩阵的第二大特征值λ2与ρ(r)的关系如下所示.
(1)
Ω(r)=Sign(λ2(r))ρ(r).
(2)
基于以上函数利用Multiwfn软件模拟的RDG等值面图以及散点图被展示在图5中[21].可以看到O1和H2之间的等值面是蓝色的,说明这里确实存在分子间氢键O1-H2…O3.同时第一激发态的等值面颜色是比基态更深的,那么说明氢键O1-H2…O3在第一激发态是增强的.从散点图也可以看出基态的尖峰值位于-0.03 a.u.处,而第一激发态的尖峰值位于-0.04 a.u.和-0.05 a.u.之间.尖峰值越负则说明氢键越强,这再一次证实了4M3HF的分子间氢键在第一激发态是增强的.

图5 4M3HF的RDG等值面和散点图
3.4势能曲线为了更加细节的描述4M3HF的质子转移路径,我们以O1-H2之间的距离作为变量扫描了4M3HF在基态和第一激发态的势能曲线,如图5所示的势能曲线是以0.03 Å为步长从0.9 Å扫描到2.15 Å获得的.从烯醇式构型到醇式构型在基态需要跨越12.91 kcal/mol的大势垒,所以可以认定基态是几乎无法发生质子转移的,而在第一激发态仅存在2.81 kcal/mol的小势垒,所以质子转移应该发生在第一激发态.同时可以看到在第一激发态中存在10.76 kcal/mol的大的反向势垒,明显比基态的2.39 kcal/mol的势垒大很多,因此4M3HF在第一激发态很难发生反质子转移,这个过程将在基态进行.4M3HF的整个质子转移过程可以总结为:在光的激发下,4M3HF的烯醇式构型吸收光子跃迁到第一激发态,之后跨越小的势垒发生质子转移形成同分异构体酮式构型,酮式构型发生辐射跃迁回到基态,最后在基态发生反质子转移回到最初的烯醇式构型.
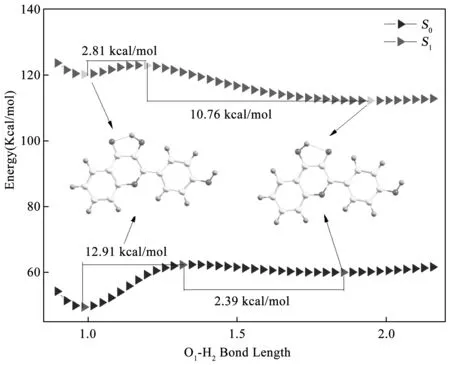
图6 基于DFT和TDDFT的方法,通过逐步加长烯醇式结构O1-H2的键的键长计算得到的势能曲线
4 结 语
通过DFT和TDDFT方法进行了4M3HF的质子转移机理的研究,运用B3LYP泛函和6-31g(d)基组优化了基态和激发态的分子结构,计算得到的吸收光谱和荧光光谱与实验结果吻合的很好.通过分析红外振动光谱,前线分子轨道以及约化密度梯度函数,我们发现分子内氢键在激发态得到了加强.最后以固定的步长构建了分子的势能曲线,通过比较势垒的大小发现质子转移是发生在第一激发态的.
猜你喜欢
数学物理学报(2022年3期)2022-05-25
数学物理学报(2022年1期)2022-03-16
数学物理学报(2021年5期)2021-11-19
食品安全导刊(2021年21期)2021-08-30
数学物理学报(2021年3期)2021-07-19
生物化工(2021年3期)2021-07-10
汕头大学学报(自然科学版)(2020年4期)2020-12-14
江苏理工学院学报(2020年2期)2020-10-23
猪业科学(2018年4期)2018-05-19
原子与分子物理学报(2015年3期)2015-11-24
