
Pituitary carcinoma: Two case reports and review of literature
2020-03-21 06:54LaiXuKaramKhaddourJieChenKeithRichRichardPerrinJianLiCampian
Lai Xu, Karam Khaddour, Jie Chen, Keith M Rich, Richard J Perrin, Jian Li Campian
Abstract BACKGROUND Pituitary carcinoma is a rare type of malignancy that can be very difficult to diagnose and treat. Many cases were diagnosed at autopsy. Delays in diagnosis often adversely impact patients' outcomes. Even with prompt diagnosis,treatment decisions remain challenging in the absence of randomized controlled trials.CASE SUMMARY We report two cases of pituitary carcinoma in men with a history of pituitary adenoma. In the first case, a 55-year-old man was initially diagnosed with pituitary macroadenoma. He underwent subtotal debulking of the tumor followed by adjuvant radiotherapy. Subsequently, he developed relapsed disease and multifocal intracranial metastases and a diagnosis of pituitary carcinoma was rendered. He passed away despite several lines of systemic therapies including temozolomide, lomustine and bevacizumab. Another 52-year-old man was diagnosed with atypical pituitary adenoma with presentation of sudden onset of vision loss in the right eye. He had recurrent pituitary carcinoma with spinal metastases, treated with surgery, radiation and temozolomide.CONCLUSION Pituitary carcinoma is a rare neoplasm with poor prognosis that is difficult to diagnose and treat. The small number of cases restricts our ability to design randomized clinical trials. Management is largely driven by retrospective studies and case series. Establishing molecular biomarkers and comprehensive genomic profiling could help in decisions about diagnosis and management of pituitary carcinoma.
Key words: Pituitary carcinoma; Diagnosis; Treatment; Pituitary adenoma;Temozolomide; Case report
INTRODUCTION
Pituitary carcinoma is a rare type of malignancy defined as non-contiguous spread of pituitary adenoma. Many cases were diagnosed at autopsy[1]. In contrast to benign pituitary adenoma, which is one of the most common intracranial tumors,representing 10%-15% of intracranial neoplasms[2], pituitary carcinoma accounts for only 0.1%-0.2% of all pituitary tumors. It can present at any age but appears more frequently in the third to fifth decade of life in patients with preexisting pituitary adenoma. It is associated with dismal prognosis and has no standardized treatment options. The median survival for patients with pituitary carcinoma, before temozolomide (TMZ) was introduced to the treatment algorithm, was about 2 years[3].This number has probably improved after the first reported case using TMZ in 2006;in one meta-analysis including a total of 54 patients, estimated 5-year survival was 57.4% for patients with atypical pituitary adenoma and 56.2% for those with pituitary carcinoma[4]. The rarity of the disease limits the design of clinical trials to study the tumorigenesis and management. Herein, we present two patients with pituitary carcinoma. Both received surgery, radiation and systemic therapies including TMZ.We hope, by sharing our experiences on this rare condition, we will help advance clinicians' recognition and management of this rare cancer.
CASE PRESENTATION
Chief complaint
Case 1:A 55-year-old male presented in September 2010 with a low testosterone level and decreased vision in the left eye.
Case 2:A 52-year-old male presented with sudden vision loss in the right eye in April 2010.
History of present illness
Case 1:The symptoms started several months prior to presentation and have been progressing gradually.
Case 2:Sudden vision loss without headache or neurological symptoms.
History of past illness
Case 1:History of clear cell renal cell carcinoma (CCRCC, stage II, T2N0M0), status post right nephrectomy in 2009.
Case 2:No past medical history.
Family history
Case 1:Unremarkable.
Case 2:Unremarkable.
Physical examination
Case 1:Unremarkable with the exception of mild decreased vision acuity in the left eye.
Case 2:Profound vision acuity in the right eye.
Laboratory examination
Case 1:Normal complete blood count (CBC), Comprehensive metabolic panel (CMP),decreased total serum testosterone.
Case 2:Normal CBC, CMP.
Imaging examination
Case 1:Magnetic resonance imaging (MRI) of the brain showed a 3.4 cm × 2.0 cm × 2.6 cm homogeneously enhancing mass in the sella with extension into the suprasellar cistern and right side of the sphenoid sinus (Figure 1A).
Case 2:MRI of the brain showed a suprasellar mass invading the optic chiasm and enveloping the carotid siphon and cavernous sinus (Figure 2A).
Case continued
Case 1:The patient was diagnosed with pituitary macroadenoma and started on hormone replacement therapy for secondary hypogonadism. He underwent left frontotemporal craniotomy with subtotal resection in February 2011. Pathologic examination yielded the diagnosis “atypical pituitary adenoma”, substantiated by a Ki-67 labeling index (LI) of 13%; the resected material showed no immunoreactivity for anterior pituitary hormones, and only weak immunoreactivity for p53 (Figure 3A).He then completed a course of adjuvant intensive modulated radiotherapy to 54 Gy,concluded in June 2011. In late 2011, the patient developed right visual loss secondary to post-radiation optic neuritis. The residual tumor remained stable (Figure 1B) on surveillance MRI studies until 2014.
Unfortunately, brain MRI in April 2014 showed significant enlargement of the tumor, with extension into the suprasellar region and left cavernous sinus (Figure 1C).He underwent transsphenoidal resection. Pathologic examination revealed histomorphologic features similar to those of the previous specimen; nuclei reactive for p53 remained rare, but the Ki-67 LI had increased to 17%. A diagnosis of recurrent atypical pituitary adenoma was rendered (Figure 3B). The patient then received adjuvant gamma knife radiosurgery to 30 Gy in 5 fractions.
In May 2015, the patient presented with confusion and focal seizures. Brain MRI/magnetic resonance angiography showed interval development of a multiloculated hemorrhagic collection around an enlarging dural enhancing lesion in the left frontotemporal convexity, adjacent to the patient’s prior craniotomy site (Figure 1D).A whole body18F-FDG-positron emission tomography/computed tomography(PET/CT) study showed increased FDG uptake within the enhancing left frontotemporal dural lesion. The patient underwent left temporal craniotomy with gross total resection of the tumor, described intraoperatively as grayish-white,gelatinous, and epidural. Histologic analysis, which included comparisons to the patient’s prior specimens, supported a diagnosis of recurrent atypical pituitary adenoma (Figure 3C). Ki-67 LI was calculated to be 25.5% and p53 stain was negative.MGMT (O-6-Methylguanine-DNA Methyltransferase) promoter methylation,evaluated upon clinical request, was not detected. The features of rapid growth, local invasion, and markedly high Ki-67 LI are considered consistent with a “clinically aggressive” adenoma; the additional finding of dissemination from the original sellar region technically completes clinicopathologic diagnostic criteria for pituitary carcinoma.
Case 2:The patient underwent right frontal craniotomy with orbitozygomatic osteotomy and subtotal resection of tumor in May 2010, at an outside institution.Histologic analysis, initially performed at Mayo Clinic in consultation, yielded a diagnosis (later confirmed at our institution) of atypical pituitary adenoma,corticotroph type, with marked pleomorphism and increased mitotic activity, ranging up to at least 5 per 10 high power fields (Figure 4A). The resection material showed no intra-tumoral Crooke hyaline change to suggest a diagnosis of Crooke cell adenoma.The patient subsequently received adjuvant Cyber Knife radiosurgery to a dose of 25 Gy in 5 fractions, completed in August 2010.
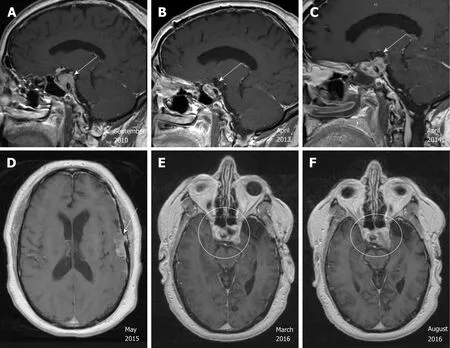
Figure 1 Radiographic images of the pituitary carcinoma discussed in case 1. A: Magnetic resonance imaging (MRI) in September 2010 shows a 3.4 cm × 2.0 cm × 2.6 cm enhancing sellar mass (arrow); B: MRI in April 2013 shows stable residual sellar lesion post-surgery and radiation (arrow); C: MRI in April 2014 shows enlarging lesion extending to the suprasellar region and left cavernous sinus (arrow); D: MRI/Magnetic Resonance Angiography in May 2015 shows multiloculated hemorrhagic collection around an enlarging dural enhancing lesion (arrow); E, F: MRI in August 2016, in comparison to MRI in March 2016, shows more enhancing solid appearing sellar mass with interval enlargement of the tumor burden along the left cavernous sinus and middle cranial fossa.
He was doing well with no evidence of progression until surveillance MRI in October 2015, which demonstrated interval development and growth of multiple right extra-axial cystic paramedian frontal lobe masses. He underwent right frontal craniotomy with resection of a 2 cm right frontal brain lesion in February 2016 at the same outside institution. Subsequent pathology review of the resection material,performed at our institution in parallel with a review of the 2010 resection material,noted strong histomorphological similarities and strong reactivity for synaptophysin,and yielded the diagnosis “metastatic atypical pituitary adenoma (atypical pituitary carcinoma)” (Figure 4B). Follow up brain MRI in June 2016 showed recurrence of the right frontal lesion as a 2.3 cm mass (Figure 2B); it also showed residual but stable enhancing tissue within the sella turcica (not shown).
In October 2016, he was referred to our institution for a second opinion on further medical management. MRI of brain and total spine (October 2016, Figure 2C and D)showed stable enhancing pituitary tumor with bilateral cavernous sinus invasion and intradural extramedullary metastatic tumor within the ventral spinal canal at T1-T3 and the T5 levels, causing severe spinal canal stenosis at T2-T3. T1-T4 decompression and tumor resection was performed on November 4th, 2016. The spinal mass was shown to be metastatic pituitary carcinoma, corticotroph subtype with reactivity for adrenocorticotropic hormone (ACTH) and synaptophysin, no significant immunoreactivity for p53, and a Ki67 LI of approximately 4.8% (Figure 4C-F);histomorphological features were considered essentially identical to those of the patient’s prior specimens.
FINAL DIAGNOSIS
Case 1 and case 2
Pituitary carcinoma.
TREATMENT
Case 1
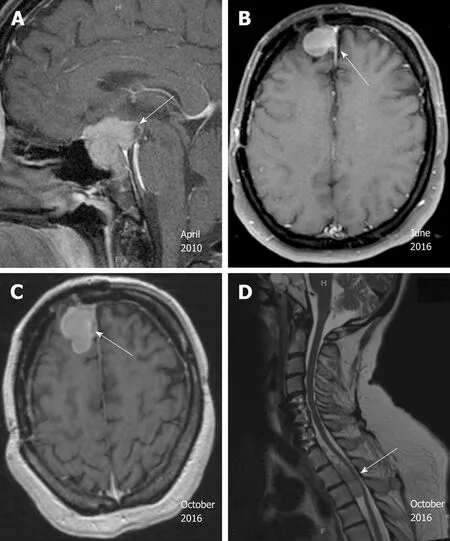
Figure 2 Radiographic images of the pituitary carcinoma discussed in case 2. A: Magnetic resonance imaging(MRI) in April 2010 shows a suprasellar mass invading the optic chiasm and enveloping the carotid siphon and cavernous sinus (arrow); B: MRI in June 2016 shows recurrence of a 2.3 cm right frontal lesion (arrow); C: MRI of brain in October 2016 shows enhancing anterior right frontal lobe mass measuring 4.1 cm × 2.1 cm (arrow); D: MRI of spine in October 2016 shows extramedullary metastatic tumor within the ventral spinal canal at T1-T3 levels (arrow,T5 lesion is not shown here).
The patient received adjuvant proton radiotherapy to 40 Gy divided in 15 fractions in July 2015. He then commenced adjuvant TMZ at 150 mg/m2for 5 consecutive days out of a 28-d cycle. The first 7 cycles were administered without major complications.Cycle 8 was delayed due to the discovery of a thyroid nodule, subsequently removed by total thyroidectomy in February 2016. Pathology revealed papillary thyroid carcinoma with metastasis to one of three lymph nodes (T3N1M0), as well as a focus suspicious for c-cell hyperplasia. He was not treated with radioactive iodine given the urgent need to resume chemotherapy. He resumed cycle 8 TMZ in April 2016. Shortly thereafter, he was noted to have a fast-growing right clavicle mass. After biopsy identified this mass as metastatic RCC, the lesion was treated with stereotactic radiation therapy with a total dose of 35 Gy delivered in 5 fractions. No systemic treatment was given for his RCC. The patient continued 2 more cycles of TMZ. Brain MRI in August 2016 revealed interval growth of the patient’s sellar mass since March 2016, with enlargement along the left cavernous sinus and middle cranial fossa(Figure 1E and F). Treatment was subsequently changed to lomustine (CCNU) at 110 mg/m2once every 6 wk. CCNU was discontinued after 2 doses due to grade 3 fatigue and a decline of Karnofsky performance status from 70% to 50%. We contemplated the ideas of comfort measures versus alternative treatment options including bevacizumab.
Case 2
After tumor resection, the patient’s course was complicated by bacterial meningitis with sphenoid erosion, cerebrospinal fluid leak and bacteremia, requiring a 3-wk hospital stay. During the admission, endocrinology was consulted for hyponatremia with workup supporting a diagnosis of syndrome of inappropriate antidiuretic hormone secretion. 8 AM cortisol level was not suppressed while the patient was on dexamethasone 2 mg every 8 h. This, together with an ACTH-immunoreactive pituitary carcinoma, raised concern for Cushing’s disease. Following the recovery and completion of steroid tapering, further endocrine workup was conducted, including low dose dexamethasone suppression test, late night salivary cortisol levels, and 24-h urine free cortisol level. He was commenced on Pasireotide for confirmed Cushing’s disease. He had secondary hypogonadism but refused testosterone replacement. He received radiation to a total dose of 30 Gy to the intracranial lesions and additional 30 Gy to the C7 though T6 spine. In January 2017, he was started on monthly high-dose TMZ chemotherapy (150 mg/m2for 5 consecutive days on a 28-d cycle). TMZ was initially planned for a total of 12 cycles. Brain MRI obtained prior to cycle 11 revealed radiographically stable disease.
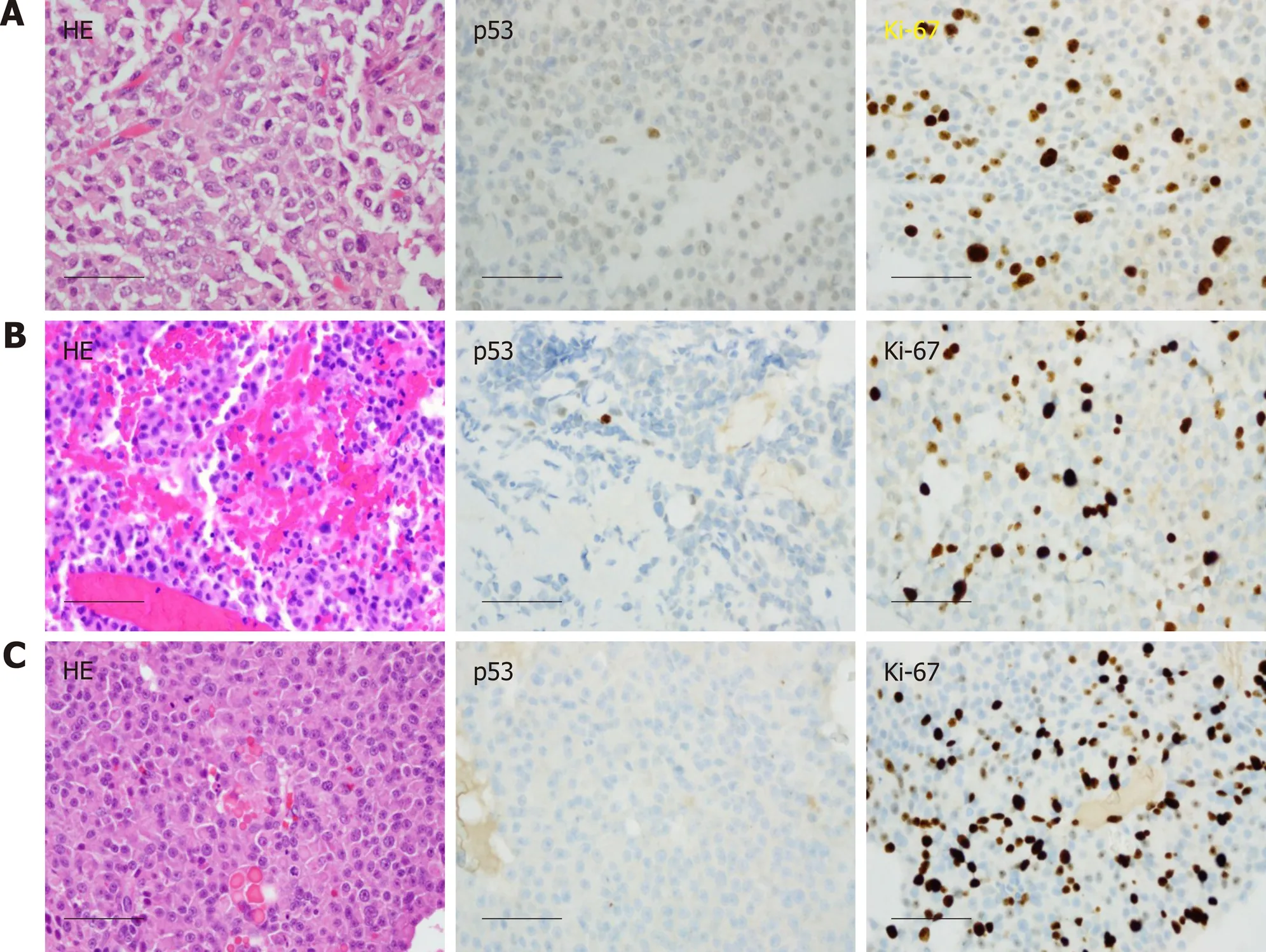
Figure 3 Histological images of the pituitary carcinoma discussed in case 1. A: "Pituitary tumor" in 02/2011 showed an atypical pituitary adenoma with fairly frequent mitotic figures (up to four per 10 high power fields), weak immunoreactivity for p53, and a Ki-67 labeling index of 13%; B: Recurrent "pituitary tumor" in 04/2014 also showed fairly frequent mitotic figures (up to two per 10 high power fields, no significant reactivity for p53, and a Ki-67 labeling index of 17%; C: The "left convexity mass" in 05/2015 showed morphological features similar to those of the previously resected tissues, but with numerous mitotic figures (at least three per most individual high power fields), a Ki-67 labeling index of 25.5%, and no reactivity for p53. Scale bar = 50 microns for all panels (400×).
OUTCOME AND FOLLOW-UP
Case 1
The patient eventually received two doses of bevacizumab (10 mg/m2, every 2 wk)and then transitioned to hospice care. The patient expired in June 2017.
Case 2
After the 10thcycle of temozolomide, the patient decided to forego further treatment and enrolled in hospice due to a decline in performance status. He expired two months later.
DISCUSSION
Classification and diagnosis of pituitary tumors
Most pituitary adenomas are slow-growing and benign, with an estimated prevalence in the general population to be 16.7%[5]. Pituitary carcinomas are rare neoplasms that are defined by disseminated disease non-contiguous with the sellar region and/or extraneural metastases. Until recently, the World Health Organization (WHO)divided pituitary adenomas into three categories: (typical) adenoma, atypical adenoma, and pituitary carcinoma; this classification system was revised in 2017[6].The term "atypical pituitary adenoma" is no longer recommended, due to poor reproducibility and predictive value. The new classification designates pituitary tumors based on adenohypophyseal cell lineages and focuses on prognostic and predictive values of histological and immunohistochemistry (IHC) markers; these IHC markers may include pituitary transcription factors, but the validity of transcription factors for this purpose is still under investigation. Tumor proliferation index and radiological invasion are still emphasized to predict aggressiveness; the use of p53 IHC, endorsed by some published reports as a predictor of aggressiveness, is considered controversial, and is now endorsed by the new WHO guidelines only for“limited” cases.
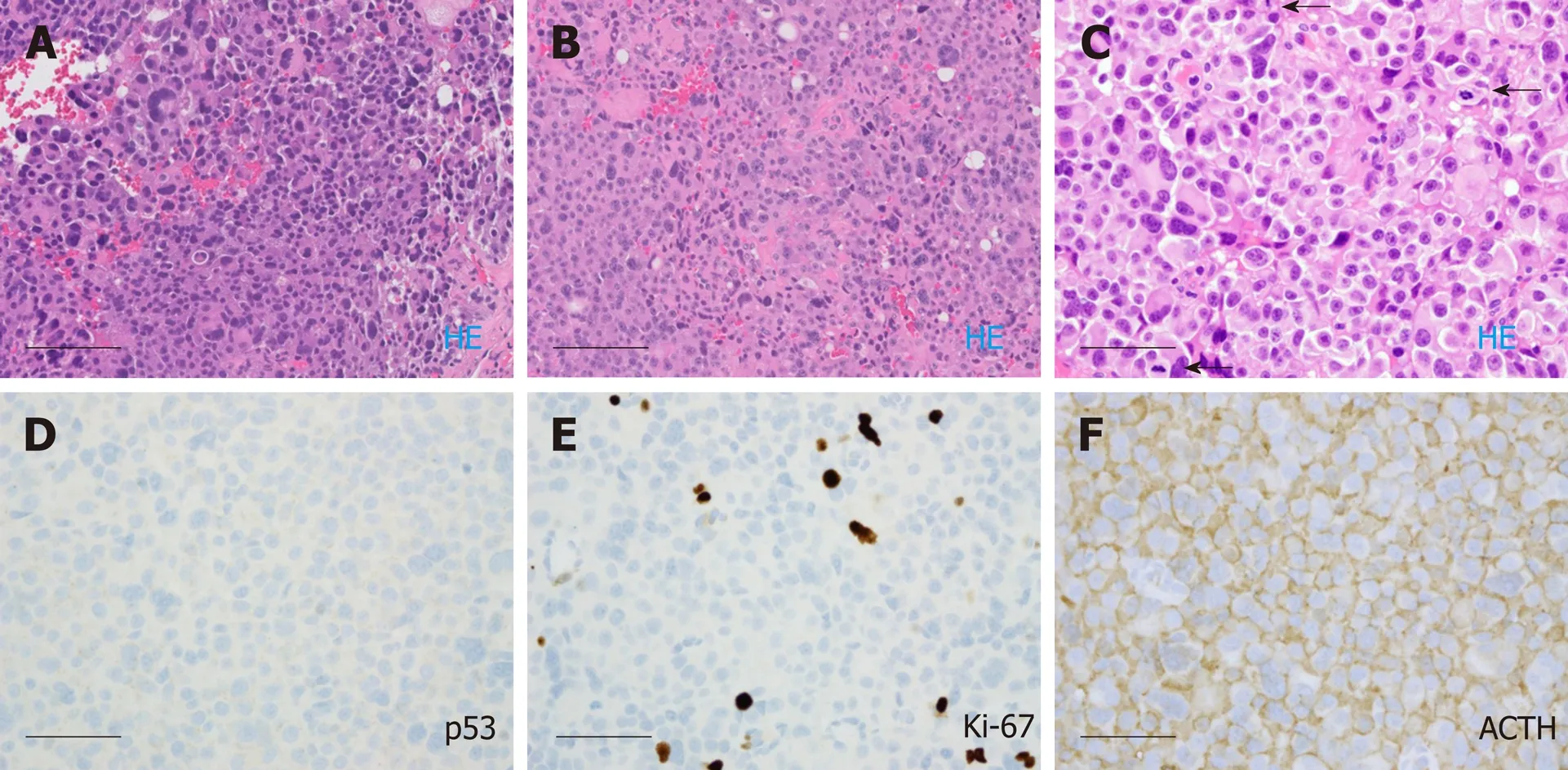
Figure 4 Histological images of the pituitary carcinoma discussed in case 2. A: "Pituitary tumor" in 05/2010 showed an atypical pituitary adenoma with marked pleomorphism and frequent mitotic figures (ranging up to five per ten high power fields); B: The "right frontal brain lesion tissue" in 10/2016 resembled the 2010 resection material from the pituitary; C-F: The spinal cord "intradural tumor" in 11/2016 resembled the previous two resection specimens, lacked significant immunoreactivity for p53, exhibited a Ki-67 labeling index of approximately 4.8%, and showed diffuse immunoreactivity for adrenocorticotrophic hormone (arrows).Scale bar = 100 microns for panels A and B (200 ×); Scale bar = 50 microns for panels C, D, E, F (400 ×).
Histomorphologically, pituitary carcinomas are intrinsically indistinguishable from pituitary adenomas. Consequently, appropriate classification of a lesion as “pituitary carcinoma” can sometimes be challenging. "Contiguous spread" versus "noncontiguous spread" does not always provide clear diagnostic distinction. As described in case 1, the involvement of dura mater could potentially result from iatrogenic“seeding” during previous resections, as opposed to pathophysiological, noncontiguous spread from the pituitary adenoma. Although we cannot exclude this potential etiology for case 1, we note that the patient's clinical course showed an aggressive pattern in parallel with the neoplasm’s elevated proliferation index.Consequently, we interpret this case as consistent with pituitary carcinoma.
Pathogenesis
The pathogenesis of pituitary carcinoma is not entirely clear. Several models have been proposed including sequential tumorigenesis and de novo transformation models[7]. In the sequential tumorigenesis model, the development of pituitary carcinoma follows a step-by-step transformation from non-adenomatous pituitary cells to adenoma cells, then aggressive pituitary adenoma and/or pituitary carcinoma.In the de novo transformation model, pituitary carcinoma cells metastasize from an aggressive pituitary adenoma that originated in normal pituitary gland. Most researchers believe in the sequential tumorigenesis model with gradual accumulation of genetic aberrations[8]; however, de novo development cannot be excluded based on current knowledge. We noticed that the Ki-67 LI in case 1 appeared to rise with every recurrence, from 13% to 17% to 25.5%, which is consistent with a step-wise transformation of the more aggressive type of pituitary neoplasm, or selection of a fast-proliferating subpopulation under treatment pressure.
The presence of coexisting RCC (clear cell type) and papillary thyroid carcinoma in the first patient certainly raised the question of an underlying germline mutation or a cancer predisposing syndrome. Unfortunately, we did not obtain consent from the patient for such molecular studies. There is literature linking pituitary carcinoma with hereditary syndrome. Succinate Dehydrogenase Subunit B (SDHB) mutation was reported in one pituitary carcinoma case[9]. SDHB germline mutation carriers have a high chance of developing paraganglioma/pheochromocytoma, and less frequently,renal clear cell carcinoma and papillary thyroid carcinoma[10-12]. Among 910 Swedish patients with germline mutations in MMR (Mismatch repair) genes (i.e., Lynch syndrome), 3 developed pituitary tumors (a higher prevalence than expected[13])including one microprolactinoma, one invasive non-secreting macroadenoma, and one pituitary carcinoma. In addition, patients with multiple endocrine neoplasia type 1 mutations are known to be at risk for pituitary adenoma as well as pituitary carcinoma[14].
Clinical presentation
Pituitary carcinomas can present clinically in three forms: As incidental findings without symptoms; as functional endocrine neoplasms, with secretory symptoms according to the type of hormone(s) secreted; or as mass lesions, with symptoms(including but not limited to headache, visual changes or diplopia[15]) often caused by the primary tumor. Most pituitary carcinomas are considered to be functional. The most common types are prolactin- (PRL) secreting and ACTH secreting carcinomas[16].In the case of PRL-secreting carcinomas, female patients may present with amenorrhea and galactorrhea; males, with erectile dysfunction[17]. Most ACTHsecreting tumors present with Cushing's disease[18], which was observed in Case 2.Pituitary carcinomas that secrete growth hormone (GH) tend to present similarly to benign GH-secreting adenomas; symptoms of acromegaly can occur[19]. Gonadotroph and thyrotroph carcinomas are the rarest and they present like benign adenomas of the same type[20,21]. There is no clear gender predilection, although some reports suggest that PRL- and ACTH-secreting carcinomas are more common in females[22].
Non- central nervous system (CNS) metastasis is more frequent than craniospinal spread in pituitary carcinoma. The most common distant metastatic location is the liver, followed by bone, lung, and lymph nodes. The interval between the initial diagnosis of pituitary adenoma and the development of invasive carcinoma is extremely variable, ranging from 4 mo to 18 years (mean, 6.6 years; median, 5.0 years)[22]. Pituitary carcinoma accounts for 0.1%-0.2% of all pituitary tumors and only a few hundred cases have been reported in medical journals[15,22,23]. Prognosis of pituitary carcinoma is generally poor, with overall survival of 2-4 years in patients with CNS metastasis, and 12 mo in those with systemic metastasis[22,24-26].
Management
Optimal treatment of pituitary carcinoma remains unknown. Multidisciplinary discussion among surgeons, radiation oncologists, medical oncologists and endocrinologists is highly recommended in the management of pituitary carcinoma.
Due to the limited number of observations, it is difficult to assess if surgery provides survival benefit. Surgical procedures are very rarely curative in pituitary carcinoma due to the local invasion of the tumor into surrounding structures.Pituitary carcinomas are usually large tumors exerting mass effects. Surgery can potentially relieve compressive symptoms and symptoms of excess hormone secretion[27]. Some may argue against surgery based on one report hypothesizing that surgical manipulation could potentially contribute to metastatic seeding[28]. In general,given the paucity of clinical trials to guide management and limited treatment options, surgery should be discussed with patients who are medically operable with tumors that are surgically resectable. The European Society of Endocrinology (ESE)guidelines recommend discussion with an expert neurosurgeon regarding surgery prior to consideration of other treatment options[29].
Radiation has been used extensively to treat primary sellar tumor and/or metastatic lesions to achieve local control of the disease. It can be administered in the adjuvant setting or to patients who are not surgery candidates[30,31]. Both fractionated external beam radiation therapy (EBRT) and stereotactic radiosurgery are effective in pituitary tumors. One retrospective study demonstrated an improved progressionfree survival (PFS) in patients with clinically non-functioning pituitary tumors who received adjuvant radiation versus those who did not (10 year PFS: 93% vs 68%, 15 year PFS: 93% vs 33%, P < 0.00005)[32]. However, at present, there are no data to suggest that radiotherapy improves survival in pituitary carcinoma[33]. Limitations of radiation mainly include long-term hypopituitarism and radiation necrosis in the temporal lobes and other nearby brain areas, especially with maximal EBRT. In addition, there is an increased risk of malignant brain tumors (RR 3.34, 95%CI: 1.06-10.6) or meningioma (RR 4.06, 95%CI: 1.51-10.9), particularly in patients treated with radiation before the age of 30 years[34]. Balancing the potential benefits and risks, ESE suggests that adjuvant radiotherapy should be considered in the setting of a clinically relevant invasive tumor remnant with pathologic markers strongly indicating aggressive behavior. Importantly, treatment decision should be discussed with an expert radiation oncologist taking into account tumor size/location, pathology, prior radiotherapy and dose[29].
There is no consensus on a standardized protocol for chemotherapy. Various combinations including TMZ, cisplatin, carboplatin, etoposide, CCNU, procarbazine,dacarbazine, paclitaxel, vincristine, methotrexate, cyclophosphamide, and doxorubicin have been used[15]. Among these, TMZ is the most widely used and appears to be effective. The first patient with pituitary tumor treated with TMZ was reported in 2006[35,36]. TMZ is a lipophilic alkylating agent that methylates guanine-rich areas of DNA and leads to DNA double strand breaks and apoptosis. It has been used extensively in the treatment of glioblastoma with success. It was reported, with confirmation by IHC and electron microscopy techniques[36], that TMZ could induce apoptosis in pituitary adenomas. In a meta-analysis of 54 patients with atypical pituitary adenoma (per previous WHO classification) and pituitary carcinoma, the objective response rate was 48.4% and the clinical benefit rate (including stable disease) was 80%[4]. There was a trend toward benefit from long-term TMZ treatment,but optimal treatment duration remains unresolved. The 5-year overall survival (OS)was 57.4% for atypical pituitary adenoma treated with TMZ and 56.2% for pituitary carcinoma[4]. OS of pituitary carcinoma prior to the introduction of TMZ was about 2 years based on historical data[3], suggesting an improvement in survival using TMZ.Negative/ low-level of MGMT IHC staining was strongly associated with response to TMZ in patients with atypical pituitary adenoma/ pituitary carcinoma[37]. The benefit of TMZ is modest but still significant given the aggressiveness and poor prognosis of pituitary carcinoma. Another French study investigated 43 patients with aggressive pituitary adenomas and pituitary carcinomas who received TMZ treatment and found a 51.2% response rate with an improved survival among responders compared with non-responders (median survival, 44 mo vs 16 mo, P = 0.002)[38].
In 2016, the ESE published a survey on 166 patients with aggressive pituitary tumors or pituitary carcinoma. 157 received TMZ as first-line chemotherapy, 7 patients received other cytotoxic or biological therapies, and 2 received peptide receptor radionuclide therapy (PRRT)[39]. The rationale of using PRRT is based on the expression of somatostatin receptors in all subtypes of pituitary adenomas,functioning and non-functioning. In 2 reports, out of 6 patients treated with PRRT,one had a partial/ complete response, one demonstrated stable disease, and the other 4 progressed on therapy[40,41]. All patients received either68Ga-DOTATATE PET-CT or octreotide scan to establish positive somatostatin receptor status prior to PRRT treatment. This, although with a small sample size, suggests potential value of PRRT in the management of pituitary carcinoma.
Additionally, TMZ in combination with capecitabine (CAPTEM) has been studied.One patient with a highly aggressive, invasive corticotroph tumor who has failed conventional medical, surgical and radiation therapies, received capecitabine 1000 mg twice daily on day 1-14 plus TMZ 200 mg/m2per day on day 10-14 administered every 28 d[42]. The patient responded well with a more than > 95% decrease in ACTH level (1874 pg/mL to 85 pg/mL) after 4 cycles of treatment, but progressed 4 weeks later. Another case series included a total of 4 patients with corticotroph pituitary tumors who were treated with CAPTEM, and showed duration of response varying between 5.5 mo to > 45 mo, with 2 of 4 patients demonstrating a complete response[43].It was proposed that sequential pretreatment with capecitabine may potentiate the cytotoxicity of TMZ, hence the unique design of this regimen is that TMZ was not started on day 1, but day 10 of the cycle. Complete response was usually not observed in patients treated with TMZ alone. Therefore, it is significant to demonstrate a complete regression of disease in 2 out of 4 patients, supporting the theoretic basis that the addition of capecitabine potentiates cytotoxicity of TMZ. In the ESE survey,there were 3 cases in which TMZ was administered as first-line treatment in combination with capecitabine. One achieved a partial response, one demonstrated stable disease and the other had progressive disease[29]. Currently, the ESE does not think there is enough evidence to support upfront TMZ in combination with other chemotherapies[29].
In patients with rapid tumor growth who have not reached maximal doses of radiotherapy, ESE recommends the "Stupp protocol", commonly used in glioblastoma.Here, TMZ is utilized as a radio-sensitizing agent at 75 mg/m2per day concomitant with 6 wk of fractionated EBRT, followed by TMZ monotherapy at 150-200 mg/m2for 5 d out of 28-d cycles. In a total of 17 cases reported in the literature, the response rate was 76%[38,44-46], higher than reported with TMZ alone, suggesting additional clinical benefit when concurrent chemoradiation is applied.
There are other chemotherapies that have been used in case reports like CCNU plus fluorouracil (5-FU), cisplatin/carboplatin plus etoposide, but these are less validated compared to TMZ containing regimens. Recently, a pediatric patient treated with the combination of carboplatin, leucovorin and 5-FU, achieved a complete radiographic response and improvement of clinical Cushing’s disease[47].
The use of targeted therapy has been explored, but findings remain inconclusive.Upregulation of Raf/MEK/ERK and PI3K/Akt/mTOR pathways and vascular endothelial growth factor (VEGF) expression have been associated with pituitary tumorigenesis. VEGF levels were higher in pituitary carcinoma than those in pituitary adenoma[48,49]. Bevacizumab, an anti-VEGF antibody, has been used in a limited number of cases to treat aggressive pituitary adenoma or pituitary carcinoma[50]. Ortiz and colleagues used bevacizumab in a patient with progressive disease on TMZ and achieved long-term disease control for at least 26 mo[51]. Bevacizumab was also administered concurrently with radiation and TMZ. Touma reported a patient who was treated post-operatively with TMZ at 75 mg/m2daily with radiation and bevacizumab at 10 mg/kg biweekly followed by 12 cycles of TMZ at 200 mg/m2for five days out of each 28-d cycle. The patient had no evidence of recurrent disease more than 5 years of follow-up[50]. Newer drugs including sunitinib (a multikinase inhibitor), Axitinib (a VEGFR inhibitor), Everolimus (an mTOR inhibitor), and Lapatinib (a EGFR/HER2 inhibitor), have been reported in cases and studied in clinical trials.
CONCLUSION
In conclusion, pituitary carcinoma is a rare neoplasm with poor prognosis that is difficult to diagnose and treat. The small number of cases restricts our ability to design randomized clinical trials. Therefore, management of this cancer has largely been dependent on information derived from case series or retrospective observations. Because clinical presentation and the interval between the diagnoses of pituitary adenoma and carcinoma are variable, a high index of suspicion is required to establish a diagnosis of pituitary carcinoma. This paper highlights the need for specific molecular biomarkers and comprehensive genomic profiling to facilitate diagnosis of pituitary carcinoma. Treatment is not standardized, and a multi-modality approach including endocrinologist, neurosurgeon, radiation oncologist and medical oncologist is highly recommended. Several regimens have shown activities in pituitary carcinoma with the most evidence supporting upfront TMZ, or TMZ plus radiation or capecitabine in selected cases. Targeted therapy and PRRT are promising areas for future research.
 World Journal of Clinical Oncology2020年2期
World Journal of Clinical Oncology2020年2期
- World Journal of Clinical Oncology的其它文章
- Can epigenetic and inflammatory biomarkers identify clinically aggressive prostate cancer?
- Objective response rate assessment in oncology:Current situation and future expectations
- Abdominal metastases of primary extremity soft tissue sarcoma:A systematic review
- Pancreatic adenocarcinoma with early esophageal metastasis:A case report and review of literature
- Metastatic clear cell renal cell carcinoma in isolated retroperitoneal lymph node without evidence of primary tumor in kidneys: A caes report