
家族性渗出性玻璃体视网膜病变一家系LRP5基因突变和临床表型分析
2020-04-07 03:35孙岩王卓实徐玲王苗苗何伟
沈阳医学院学报 2020年1期
孙岩,王卓实,徐玲,王苗苗,何伟
(沈阳何氏眼科医院遗传门诊,辽宁 沈阳 110033)
家族性渗出性玻璃体视网膜病变 (familial exudative vitreoretinopathy,FEVR)是一种临床和遗传上异质性很强的一类疾病,它是由Criswick和Schepens[1]于1969年首次发现和提出的,此后国内外对FEVR的研究逐渐深入。FEVR临床异质性表现可以从无任何症状到视网膜完全脱离,同一家庭的不同先证者之间也可能存在临床症状差异,甚至单一先证者的双眼表型也可能不同,这给疾病的诊断和干预带来了很大的困难。该疾病的典型临床表现是双眼周边视网膜的血管发育异常,伴有新生血管、视网膜出血、视网膜褶皱、玻璃体渗出、玻璃体和视网膜粘连等,发病严重者会导致失明[2-3]。根据临床表现和眼底荧光血管造影(FFA)诊断,结合基因检测,可以帮助辅助诊断FEVR,并了解整个家族的基因突变携带情况[4-5]。先前的研究发现FEVR具有多种遗传模式,包括常染色体显性遗传、常染色体隐性遗传和X连锁隐性遗传[6],其中常染色体显性遗传是FEVR中最常见的形式[7]。迄今为止发现,FZD4、LRP5、KIF11、NDP、ZNF408基因均与 FEVR 有关[8-12],其中FZD4、LRP5、KIF11基因遗传模式是常染色体显性遗传,并在韩国人群中致病突变检出率高,合计占比 35.3%,而LRP5基因突变占比5.6%[13],但是LRP5基因在中国人群中报道的突变频率较低。有研究发现LRP5基因参与Norrin信号蛋白分子介导的Wnt/β-catenin信号通路,该通路在组织发育和细胞分化中起着非常重要的作用,不仅参与细胞的增殖、凋亡、分化、血管发育和胚胎发育,而且与血管生成密切相关[14]。Wnt/βcatenin信号通路在生物进化中高度保守,并在许多生命活动中起着至关重要的作用,临床上可以表现出胚胎眼底新生血管形成,这种病理变化是导致FEVR的主要原因[15]。基于遗传因素在FEVR的发病机理中的重要影响因素,以及早期临床确诊的困难性,有必要寻找其他致病基因或致病突变,并深入研究FEVR的发病机理,为该类疾病的早期干预和临床治疗提供必要的研究基础。
1 资料与方法
1.1 研究对象 将2019年1月在沈阳何氏眼科医院确诊的1例FEVR先证者家系纳入本研究,其中男1例,女3例。FEVR的诊断标准:(1)眼底检查显示周边视网膜无血管; (2)FFA显示周边视网膜无血管,可发现不同程度的视网膜下液或视网膜脱离;(3)无早产、低出生体重和吸氧史的儿童;(4)FEVR家族史; (5)排除Norrie病和早产儿视网膜病变。先证者及其家系成员由同一名视网膜专家进行评估[16]。排除标准: (1)不能配合完成相关检查者; (2)无法判断发病原因是由遗传因素引起的; (3)无家系血样可以进行基因验证者。本研究经沈阳何氏眼科医院伦理委员会的批准 (批文号:IRB(2016)K001.01),所有先证者及其家系成员均签署知情同意书。
1.2 获取先证者和家系成员相关病史及进行相关物理检查 先证者和家系成员均通过标准眼科临床检查,以明确诊断,并排除其他非遗传因素引起的眼部疾病。病史包括基础个人信息 (包括性别、年龄、家庭情况、籍贯、民族等)、主诉、现病史 (发病情况、就诊情况、用药情况、并发症种类和时间等)、既往史 (其他器官病变、基因检测史等)、遗传史、手术和药物史、婚育史等。物理检查包括视力检查和矫正视力、非接触性眼压、B超 (天津迈达,型号 ODM-2200)、眼底照相(Topcon,型号TRC NW-300)、光相干断层扫描(卡尔蔡司,型号Cirrus HD-OCT 5000)和眼底荧光造影 (海德堡,型号Spec-KT)检查。
1.3 高通量二代基因测序
1.3.1 样本采集 常规采集符合要求的先证者及家系成员肘部静脉血5 ml,ETDA抗凝,-80℃保存。
1.3.2 靶向基因捕获和变异解读 (1)DNA提取:Qiagen试剂盒 (荷兰Qiagen公司,型号Flexi-Gene)提取先证者及家系成员外周血的DNA,Qubit荧光仪 (赛默飞中国,型号Qubit 4)检测DNA浓度,琼脂糖凝胶电泳仪 (赛默飞中国,型号Fastruler SM1103)检测DNA完整性;(2)文库制备:参考华大基因的BGISEQ-500的文库构建指导手册进行;(3)上机测序:质控合格的文库采用深圳华大基因股份有限公司BGISEQ-500 PE50+10定制基因捕获芯片的NGS程序上机测序,共检测792个眼科目标基因编码外显子、±3 bp的内含子区域和非编码区域 (UTR)[17]。按照制造商的标准流程,通过PCR扩增DNA片段,并与特殊设计的DNA捕获探针杂交,将洗脱的DNA片段再次扩增,并使用 Illumina HiSeq 2500 Platform(美国Illumina公司)测序系统进行NGS检测。
1.3.3 序列和生物信息学分析 从测序仪获取原始数据 (FASTQ数据),对原始FASTQ数据进行质量控制,去除低质量数据,利用BWA(BWAMEM,版本0.7.10)软件,使用hg39人类参考基因组参考序列进行比对,基于Picard去除重复序列,使用基因组分析工具套件 (GATK,版本3.3)对数据进行突变检测,使用频率数据库db-SNP、千人基因组数据库 (G1000)、ESP6500数据库、ExAC数据库以及BGI内部数据库进行频率注释,去除次要等位基因频率 (MAF)>0.1%的变异位点;使用人类基因组变异协会 (HGVS)规则对突变进行标准命名;使用OMIM、HGMD等疾病数据库进行突变及疾病注释。
1.3.4 检测结果解读筛选候选基因致病变异(1)筛选、过滤:按照注释标准 (MAF≤0.05%,G1000、 dbSNP、 ESP6500、 ExAC≤0.01) 去除数据库中正常人群携带频率较高的非致病性突变,去除内含子区、UTR区和基因间突变,去除同义突变等突变类型; (2)有害性评估:通过SIFT、PolyPhen等软件预测,去除预测为良性或中性的突变,筛选出保守性强并且具有危害性的突变,根据美国医学遗传学和基因组学学会 (ACMG)的标准和指南评估变异的有害性; (3)家系共分离验证:通过家系样本Sanger验证,确认致病基因突变在家系成员之间的遗传方式和临床表型对应关系。 (4)致病性突变分类:致病性分类按照《ACMG遗传变异分类标准与指南》提供的规范,使用特定标准术语来描述,包括 “致病的”、 “可能致病的”、“临床意义未明”、“可能良性的”和“良性的”。
1.3.5 Sanger验证 利用Primer3设计所有聚合酶链反应引物进行验证。本研究4例家系成员均进行Sanger测序,用于验证所鉴定的突变,并通过家系共分离验证。
2 结果
2.1 测序质量结果 该家系目标序列平均测序深度为112.47,目标序列覆盖率平均99.94%。平均99.98%的目标序列覆盖率>10×,平均97.91%的目标序列覆盖率>30×,测序深度和平均覆盖率均符合检测要求。
2.2 家系临床表型情况 本研究4例家庭成员中FEVR患者3例,正常表型家系成员1例,见图1。罹患FEVR的3例家系成员最佳矫正视力均低于指数视力,且均于出生时发现特征性临床表征,其中先证者及其姐姐和父亲分别同时伴发右眼牵拉性视网膜脱离、永存原始玻璃体增生症 (persistent hyperplastic primary vitreous,PHPV)和核性白内障,见表1。全视野眼底照相和FFA检查显示先证者的临床表型符合FEVR特征,但先证者姐姐和父亲因为并发双眼眼球震颤和核性白内障,仅留取部分眼底照相。先证者FFA表现为双眼周边视网膜血管分支增多,分布密集,呈现典型的“柳枝”样形态,伴颞侧牵引性视网膜脱离和视网膜内/下渗出,伴镰状视网膜固定褶皱。但是因同一基因突变致病的先证者姐姐却出现左眼PHPV,表现为后极部视网膜表面一灰白色条索样增殖膜自视盘处向颞上方周边视网膜延续,增殖膜下方视网膜表现出脱色素,伴视网膜色素上皮层萎缩及色素团块析出。先证者父亲因为双眼核性白内障,无法获得清晰的眼底检查报告。先证者母亲眼底表现正常,未发现周边视网膜血管渗漏和新生血管增殖。见图2、图3。

图1 FEVR先证者家系图及基因携带情况
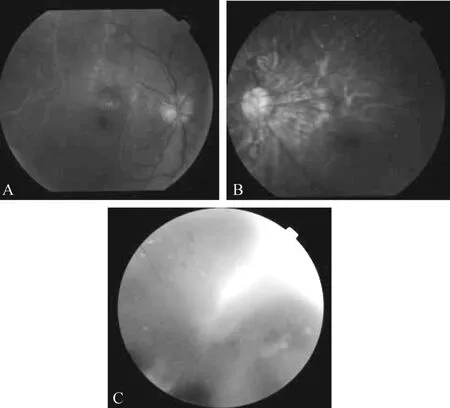
图2 家系眼底照相图
2.3 基因检出情况 本研究发现了LRP5基因的一个未报道的缺失突变c.2013delC,该突变位于LRP5基因的9号外显子,呈常染色体显性遗传,通过家系共分离和Sanger验证,被确认为该家系FEVR的疑似致病突变,见图4。

表1 FEVR家系临床信息
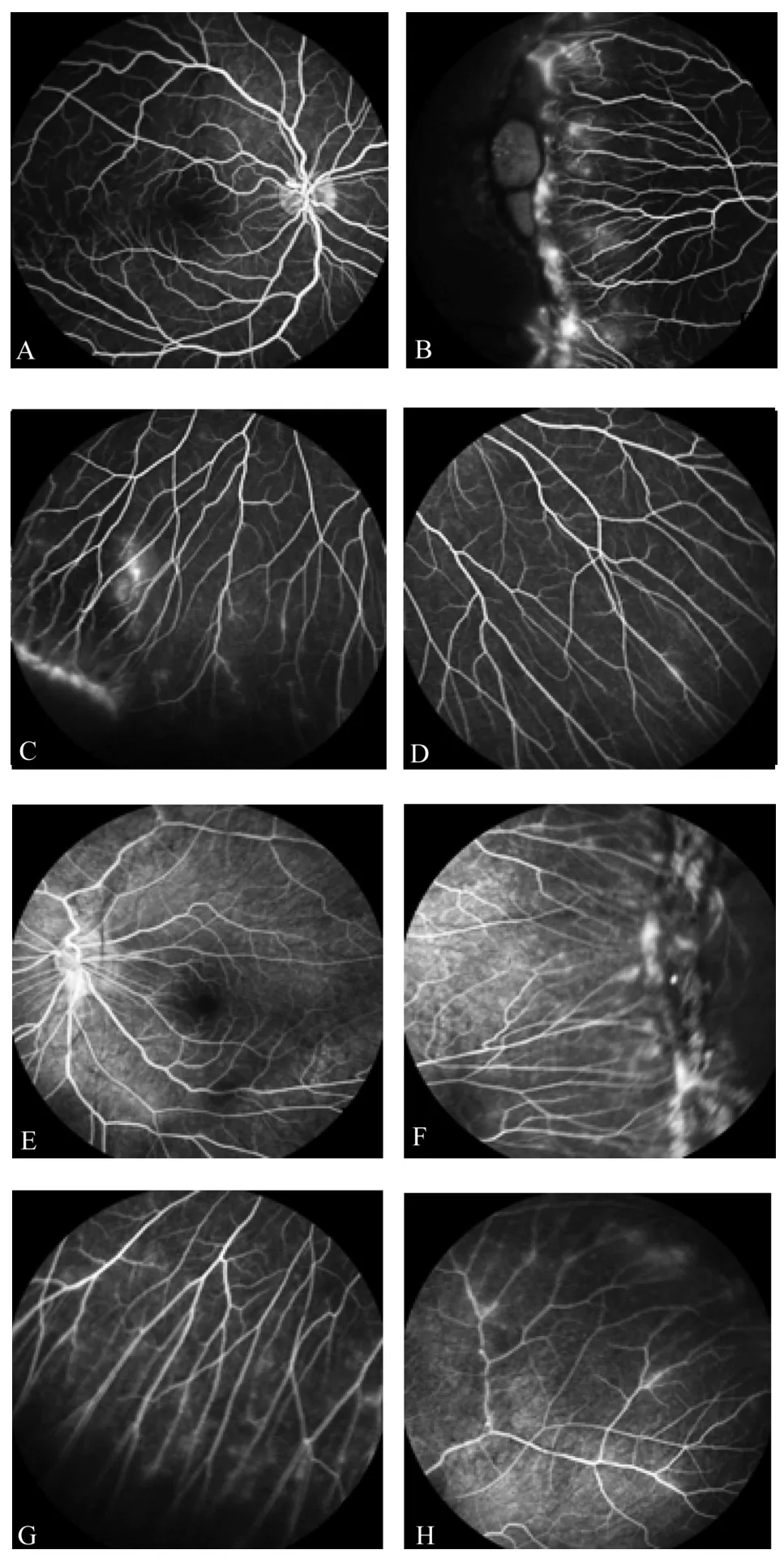
图3 先证者FFA图
3 讨论
FEVR是一类遗传和临床异质性非常强的遗传眼病,主要表现为周边视网膜新生血管和纤维膜增殖。患者可能在发病早期无明显的症状和体征,而在后期可能出现严重的玻璃体出血和牵拉性视网膜脱离,导致视力下降甚至失明。本研究收集的FEVR家系被检出来自于低密度脂蛋白受体相关蛋白 (LRP5) 的基因突变。Toomes等[18]在一显性遗传的FEVR家系中发现了LRP5基因突变,后续又有其他研究证实了FEVR家系的LRP5突变[19-20]。LRP5基因定位于人染色体 11q13.4,全长136.6 kb[21],LRP5跨膜蛋白能与FZD4基因编码的Frizzled4蛋白形成受体复合物,参与活化Wnt/β-catenin信号通路。LRP5基因突变最初是在患有隐性遗传的骨质疏松伴假性神经胶质瘤综合征 (osteoporosis pseudoglioma syndrome, OPPG)的患者中发现[22],目前认为该病与FEVR是同一种疾病,该病的典型特征是儿童骨质疏松和FEVR的眼部表现。主要致病机制可能是由于LRP5基因不仅参与视网膜血管发育,还在嗜铬样细胞中通过抑制血清素的合成来促进成骨细胞和骨形成中起到作用,导致了大多数OPPG患者出现严重的双侧视网膜发育不良或先天盲,同时伴有骨密度明显减低。

图4 家系Sanger验证结果
本研究首次报道了LRP5基因的一个未报道的缺失突变c.2013delC,该突变位于LRP5基因的9号外显子中,呈常染色体显性遗传,该缺失移码突变引起阅读框变化,使编码蛋白质从突变位点开始氨基酸序列发生改变,导致LRP5基因分别在突变位点后的第26个氨基酸位置发生截短终止,进而损害编码蛋白质的功能。通过UCSC(http://genome.ucsc.edu/)在线工具对突变位点的氨基酸进行保守性分析,显示c.2013delC突变位点在人类、恒河猴、小鼠、狗、大象、家鸡等脊椎动物中相对保守,提示这些突变对LRP5基因功能可能会造成严重影响。
FEVR的早期诊断决定了疾病的预后,由于新生血管形成对视觉结果的影响,早期诊断可以保持更好的视力。目前主流的诊断主要依赖于FFA的精确表现和临床眼底的典型症状,对于活动性病变,临床主张预防性治疗。有研究表明,基因突变导致的无症状FEVR家庭成员中有58%具有一期和二期FEVR的临床特征和血管造影证据[16]。本研究认为,对于未表现出临床症状的FEVR基因突变家系成员,荧光素眼底血管造影联合基因测序技术与单纯通过FFA和眼底表现做出的诊断相比,可以显著提高FEVR的早期诊断率,进而及时干预病程进展,获得更佳视力。
Robitaille等[24]发现FZD4基因突变可能会导致与PHPV相似的临床表现,Tang等[25]也发现在FEVR病例家系中存在PHPV患者病例,说明PHPV和FEVR的临床表型可能有重叠。本研究中的家系成员临床表现与之前的报道相似。先证者表现为典型的双眼周边视网膜血管分支增多,分布密集,呈现典型的 “柳枝”样形态,伴颞侧牵引性视网膜脱离,和视网膜内/下渗出,伴镰状视网膜固定褶皱。但是同样基因突变致病的先证者姐姐却发现左眼PHPV的特征表现,表现为后极部视网膜表面一灰白色条索样增殖膜自视盘处向颞上方周边视网膜延续,增殖膜下方视网膜表现出脱色素,伴视网膜色素上皮层萎缩及色素团块析出。上述结果表明,具有相同基因突变的多个个体可以表现出不同的临床表型,反映出遗传性FEVR的高度临床异质性,这对于进一步了解基因型-表型非常重要。
综上所述,本研究扩大了FEVR的致病基因突变谱,并展示了FEVR基因型与临床表型的复杂性。本研究表明FEVR具有高度的临床异质性,特别是对于没有家族病史或非典型症状的患者,在临床诊断和遗传咨询过程中不应该基于基因型或表型单方面进行分析,应充分考虑基因型与表型之间的关联,在为FEVR患者的家庭成员提供遗传咨询服务时,要进行全面的眼底检查。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
临床输血与检验(2022年3期)2022-06-22
实用肝脏病杂志(2022年2期)2022-03-21
诊断学(理论与实践)(2020年1期)2020-04-28
水产科学(2020年2期)2020-03-20
小学生导刊(2018年13期)2018-06-29
森林工程(2018年1期)2018-05-14
中学生理科应试(2017年6期)2017-09-27
医学研究杂志(2015年12期)2015-06-10
