
碳化终温对β-Mo2C催化喹啉加氢脱氮性能影响
2020-05-06 03:27邱泽刚马少博李志勤
燃料化学学报 2020年3期
邱泽刚, 李 侨, 马少博, 李志勤
(西安石油大学 化学化工学院, 陕西 西安 710065)
煤焦油加氢制燃料油品已形成较大的产业规模。但是,燃料油品价格受石油价格波动影响显著,新能源的快速发展对油品市场的潜在影响也将逐渐显现,单一的制取燃料油品的生产模式经济效益日趋走低,长远来看难以持续,这使得对发展产品多元化、高值化的煤焦油加工技术的需求越来越迫切。煤焦油转化为燃料油品的过程包括硫、氮、氧和重金属等有害及污染性物质的脱除,烯烃、部分芳烃饱和与大分子裂化等过程。煤焦油中氮含量(0.45%-1.30%)一般高于硫(0.29%-0.40%)[1],且在硫、氮和氧三类杂原子中,氮加氢脱除反应的难度最大,硫脱除难度居中,氧脱除的难度最小[2],故在煤焦油加氢过程中,一般氮、硫被脱除时氧也会被脱除[3-6]。因此,加氢脱氮(HDN)是煤焦油加氢过程中的关键问题[1]。必须指出,HDN也是石油加工的关键过程之一[2,7]。
加氢脱氮(HDN)必须在催化剂作用下进行,当前煤焦油加氢装置中使用的是硫化态加氢精制催化剂,与石油加氢过程中使用的催化剂类似,这些催化剂的主要作用是尽可能地脱除油品中的氮,因此,主要关注的是加氢脱氮的活性,而并未考虑脱氮过程中是否造成高价值结构单元的破坏。实际上,这些催化剂会在很大程度上破坏煤焦油中具有较高潜在价值的芳环[3,4]。很显然,若使煤焦油加工技术向产品多元化、高值化的方向发展,需在HDN过程中尽可能保持高价值的芳环不被破坏。这对煤焦油HDN催化剂提出了不同以往的新的要求,即在加氢脱氮的同时尽量减少芳环的破坏。需指明的是,减少芳环的破坏将会自然的降低加氢过程的氢耗。
煤焦油中含氮化合物主要是喹啉、吡啶,吲哚、咔唑及它们的衍生物,还有少量脂肪胺[1,3],其中,喹啉因同时含有芳环和氮杂环,是最具代表性的模型分子。因此,本研究选取喹啉作为模型物进行HDN催化剂研究。喹啉是研究石油馏分HDN常用的模型分子,已有较多相关研究报道,涉及到催化剂载体及活性组分、热力学、动力学及催化反应机理等方面[8-14]。喹啉在催化剂上的HDN反应,可分为两种典型反应途径[3,8]:(Ⅰ)部分加氢途径,THQ1—OPA—PB和(Ⅱ)完全加氢途径,DHQ—PCHA—PCH。
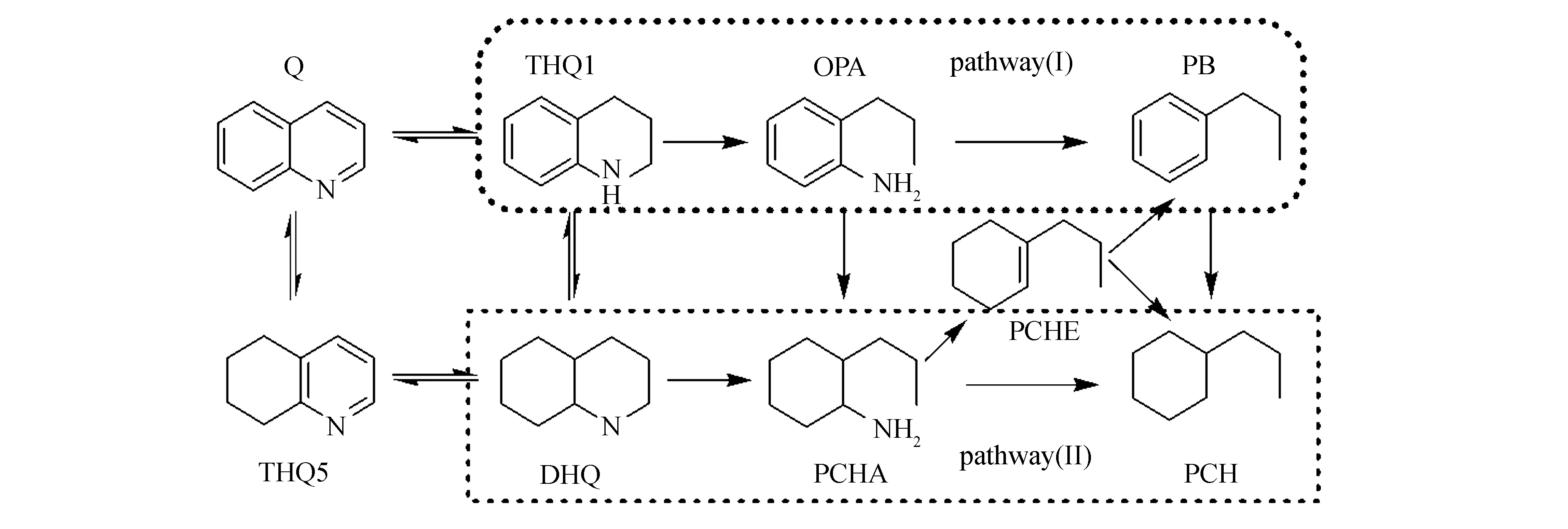
图1 喹啉的HDN反应网络及反应途径示意图
在对喹啉HDN研究中,以金属催化剂为主,包括硫化物和非硫化物,非硫化物主要是磷化物、氮化物和碳化物。负载的过渡金属硫化物(硫化态的Ni、Mo或W等)[8-10]是最为经典的催化剂,磷化物喹啉HDN活性最为突出,但硫化物和磷化物均明显倾向于完全加氢途径(Ⅱ)[8,11,12]。与硫化物和磷化物相比,碳化钼显示出了对于PB的高选择性(表明芳环得以保持),即对路径(Ⅰ)的高选择性。Schlatter等[13]报道了Mo2C/Al2O3和Mo2N/Al2O3的喹啉HDN性能,两者活性与商业Ni-Mo/Al2O3相当,但对产物PB选择性显著高于Ni-Mo/Al2O3,其中,尤以Mo2C/Al2O3选择性最高,在相同脱氮率下,尤其是在处理氮含量高于硫含量的原油时,Mo2C/Al2O3具有比Ni-Mo/Al2O3低得多的氢耗。Dolce等[14]报道显示,Mo2C/Al2O3对苯类产物的选择性要比Ni-Mo/Al2O3高出约20%,证实了Schlatter等[13]的研究结果。然而,碳化钼对路径(Ⅰ)的高选择性在之后多年都没有引起太多重视。因为对于石油的加氢脱氮而言,高活性是首要的需求,且石油中氮含量一般都会低于硫含量,这与高芳烃含量且氮含量显著高于硫含量的煤焦油有显著的差别。总体而言,关于HDN过程中碳化钼活性相、活性位和相关机理的信息仍非常有限。需指明的是,碳化钼(Mo2C)由于其类铂电子结构在加氢脱硫、加氢脱氧、烷烃异构化和电解析氢等反应过程中都显示了高效、稳定的催化性能[15,16]。实际上,碳化钼存在两种较稳定的晶相,六方晶相β-Mo2C和立方晶相α-MoC[17]。2018年,Sebakhy等[18]在较低温度下(500 ℃)利用程序升温还原(TPR)制备了立方相α-MoC0.5和六方相β-Mo2C,并使用密度泛函理论(DFT)计算了立方相向六方相的转变。将其用于1-辛烯和甲苯加氢,发现两种相态具有相似的活性,甲苯的损失非常小,且β-Mo2C具有更优异的低压加氢能力。
本研究以CH4/H2为碳源制备了一系列碳化钼催化剂,通过改变碳化终温改变碳化钼的结构性质,进而研究β-Mo2C结构性质变化对喹啉加氢脱氮过程的影响,为进一步研究碳化钼活性相、活性位和相关机理,发展低芳环破坏、低氢耗的焦油加氢脱氮催化剂打下基础,也为煤焦油向芳烃和特种油品的转化以及性质类似的煤基液体(如煤液化油)的转化提供参考。
1 实验部分
1.1 试剂
钼酸铵(分析纯,天津市科密欧试剂有限公司),喹啉(分析纯,上海麦克林生化科技有限公司),正庚烷(分析纯,天津市科密欧试剂有限公司),CH4、H2和N2(99.99%,腾龙化工气体公司)。
1.2 催化剂的制备
采用程序升温直接还原碳化法制备碳化钼:称取一定量的钼酸铵,在马弗炉中于500 ℃下,焙烧4 h,压片、筛分,即得到前驱体三氧化钼。将三氧化钼于真空加热炉中进行程序升温还原碳化。碳化前先抽真空再通入氮气吹扫。混合气(CH4/H2)的总体积空速为6000 h-1,碳源浓度为20%,以10 ℃/min升温至350 ℃,再以1 ℃/min升温至设定终温(640-720 ℃),恒温3 h。然后室温下以O2/N2气流钝化3 h,制得碳化钼催化剂。
不同碳化终温的碳化钼催化剂分别记为Mo2C-640、Mo2C-660、Mo2C-680、Mo2C-700和Mo2C-720;反应后的催化剂分别记为Mo2C-640af、Mo2C-660af、Mo2C-680af、Mo2C-700af和Mo2C-720af。
1.3 催化剂的表征
X射线衍射(XRD):日本岛津的XRD6000型衍射仪,CuKα光源(λ=0.154 nm),10°-90°扫描,步长0.02,扫描速率10(°)/min;
N2吸附-脱附:美国麦克仪器的ASAP 2460系列全自动比表面积与孔隙度分析仪,在350 ℃下预处理4 h。
扫描电镜(SEM):美国FEI的Quanta600FGG型扫描电子显微镜,工作电压5 kV。
透射电镜(TEM):美国FEI的Tecnai G2 F20型场发射透射电子显微镜,使用铜网进行制样,工作电压5 kV。
X射线光电子能谱(XPS):日本ULVAC-PHI公司PHI5000VersaprobeIII型能谱分析仪,X光源为AlKα(hv=1486.6 eV),用污染碳(C 1s=284.6 eV)对结合能进行校正。
拉曼光谱(Raman):法国HORIBA的LabRam ARAMIS全自动拉曼光谱仪,激光波长为532 nm,电荷耦合器件(CCD)室温探测器,功率为7 mW。
1.4 催化剂的性能评价
采用高温高压固定床,反应管的长度600 mm、内径5 mm、外径14 mm,催化剂装填量为1.0 g。在压力4.0 MPa、温度300 ℃的氢气气流下活化1.5 h,再升温至反应温度340 ℃。然后浓度为2%喹啉正庚烷溶液经高压液相泵进入反应系统,液时空速12 h-1,氢油比500∶1,持续3 h后,进行取样,使用福立公司的GS9790Plus气相色谱仪进行分析,采用内标法定量(以正癸烷为内标物)。
2 结果与讨论
2.1 催化剂的性能评价
2.1.1 碳化终温改变时碳化钼喹啉HDN性能
图2为不同碳化终温对碳化钼催化剂性能的影响。由图2可知,碳化钼催化剂的不同碳化终温对喹啉加氢脱氮反应性能有较大的影响。随着碳化终温的升高,喹啉加氢脱氮反应的转化率和脱氮率均呈现先上升后下降的趋势(图2(a)),且增加幅度很大,Mo2C-680(碳化终温为680 ℃)比Mo2C-640和Mo2C-660的喹啉加氢脱氮转化率分别提高了4.0%和1.5%,脱氮率分别提高了约11.0%和9.0%。与Mo2C-680相比,Mo2C-700和Mo2C-720上喹啉加氢脱氮转化率分别降低了1.0%和1.5%,脱氮率分别降低了3.0%和3.5%。芳烃类和环烷烃类产物选择性也呈先升后降的趋势(图2(b)),但两者变化幅度有显著差异。与Mo2C-660相比,Mo2C-680上芳烃类选择性提升了7.0%,而环烷烃类选择性仅提升0.6%。Mo2C-700上芳烃类选择性比Mo2C-680仅降低0.6%,环烷烃类降低2.0%。上述数据表明,碳化终温由660 ℃升至680 ℃,提升的转化率大部分贡献给了芳烃类产物(部分加氢途径)。显然,通过改变碳化终温,可以显著改变碳化钼喹啉加氢脱氮活性和对芳烃类产物选择性。当反应温度340 ℃、压力4 MPa、液时空速12 h-1、氢油比500∶1时,Guo等[19]采用MCM-41负载的Ni-W基催化剂对喹啉加氢脱氮性能的影响研究,发现反应温度为350 ℃时,转化率可达97%,但脱氮率只有32.6%。而本研究中程序升温碳化制备的所有催化剂在340 ℃时,喹啉转化率均高于96%,脱氮率均大于88%,表明该催化剂的加氢脱氮性能较佳。

图2 不同碳化终温对碳化钼催化剂性能的影响
2.1.2 反应条件改变时碳化钼喹啉HDN性能
不同反应条件(包括反应温度、压力、氢油比和空速)下碳化钼催化剂喹啉HDN性能见图3。随着反应条件的变化,总的HDN速率(或总的脱氮率)均出现一个最高点,且呈现先增加后减小的趋势。就温度因素而言,随着反应温度升高,氢解(C-N键断裂)速率常数提高,有利于HDN速率提高;而同时加氢反应平衡常数会下降,导致总的HDN速率下降。当温度达到340 ℃时,反应处于最有利阶段,当温度低于340 ℃时,HDN主要受动力学控制;当温度高于340 ℃时,该反应主要受热力学控制。由图3可知,反应温度、氢油比、液时空速对芳香族类的选择性影响不大,但压力影响较为显著,随着反应压力的提高(≤4 MPa),芳香族类化合物的选择性增大,当反应压力高于4 MPa时,芳香族类化合物的选择性降低,但环烷烃类的选择性一直呈现增加趋势,这表明进一步提高压力有利于芳环的饱和。反观环烷烃类选择性,除空速外,反应温度、压力和氢油比均对其影响较大,反应温度、氢油比增大,环烷烃类选择性均是先增加后减小,且增加幅度较大,这与转化率和脱氮率的变化趋势一致,表明改变反应温度、压力和氢油比可以提升转化率,且主要有利于促进环烷烃类的生成。黄澎[20]考察了SBA-15负载磷化镍催化剂对喹啉加氢脱氮反应网络的影响,发现喹啉加氢脱氮产物主要为丙基环己烷(91.6%)和丙苯(4.9%)。尹海亮等[21]研究了乙二醇对磷掺杂NiMo/Al2O3催化剂加氢脱氮性能的影响,发现喹啉加氢脱氮主要产物为丙基环己烷(88.88%)、丙基环己烯(5.07%)和2-丙基苯胺(5.72%)。本研究制备的所有催化剂在考察的反应条件下,芳香族类的选择性均在30%以上,这表明碳化钼催化剂更有利于芳香类化合物的生成。而产物中出现苯、甲苯和乙苯,显示β-Mo2C具有一定的使芳环侧链C-C键断裂的性能。
2.2 催化剂的表征
2.2.1 催化剂的XRD表征
图4为MoO3和不同碳化终温碳化钼催化剂反应前后XRD谱图,由图4(a)可知,MoO3在12.79°、23.35°、25.70°、27.15°、34.36°、39.66°、46.24°和49.71°处出现α-MoO3的特征衍射峰。不同碳化终温碳化后,Mo2C-640、Mo2C-660、Mo2C-680、Mo2C-700和Mo2C-720均于34.51°、38.02°、39.45°、52.29°、69.76°、74.97°和75.98°处出现典型的β-Mo2C特征峰,但均未检测到钼氧化物及其他物相,这显示所有催化剂体相为β-Mo2C,也即是热力学稳定的六方结构(HCP)[22]。随着碳化终温的不断升高,碳化钼的特征峰强度出现小幅增强,而半峰宽稍减小,表明结晶度略有增强。
由图4(b)可知,反应后的催化剂均未出现钼氧化物及其他物相特征峰,与反应前相比,物相未发生变化,表明反应过程中各催化剂体相较为稳定,但β-Mo2C特征衍射峰峰强度均出现一定降低,显示结晶度略有降低,碳化终温越高,β-Mo2C特征衍射峰峰强度降低幅度越小,显示碳化终温越高越有利于物相的稳定。
由XRD分析结果可知,各碳化终温下制得的催化剂体相均为β-Mo2C,随碳化终温升高,催化剂体相物相保持不变,结晶度和物相稳定性均呈略增强的趋势。前文可见,随碳化终温升高,各催化剂上喹啉HDN反应的转化率和脱氮率变化幅度大,且均呈现先上升后下降的趋势(图2),这与催化剂体相、结晶度、物相稳定性变化趋势完全不符,因此,体相不是造成催化剂喹啉加氢脱氮性能差别的原因。
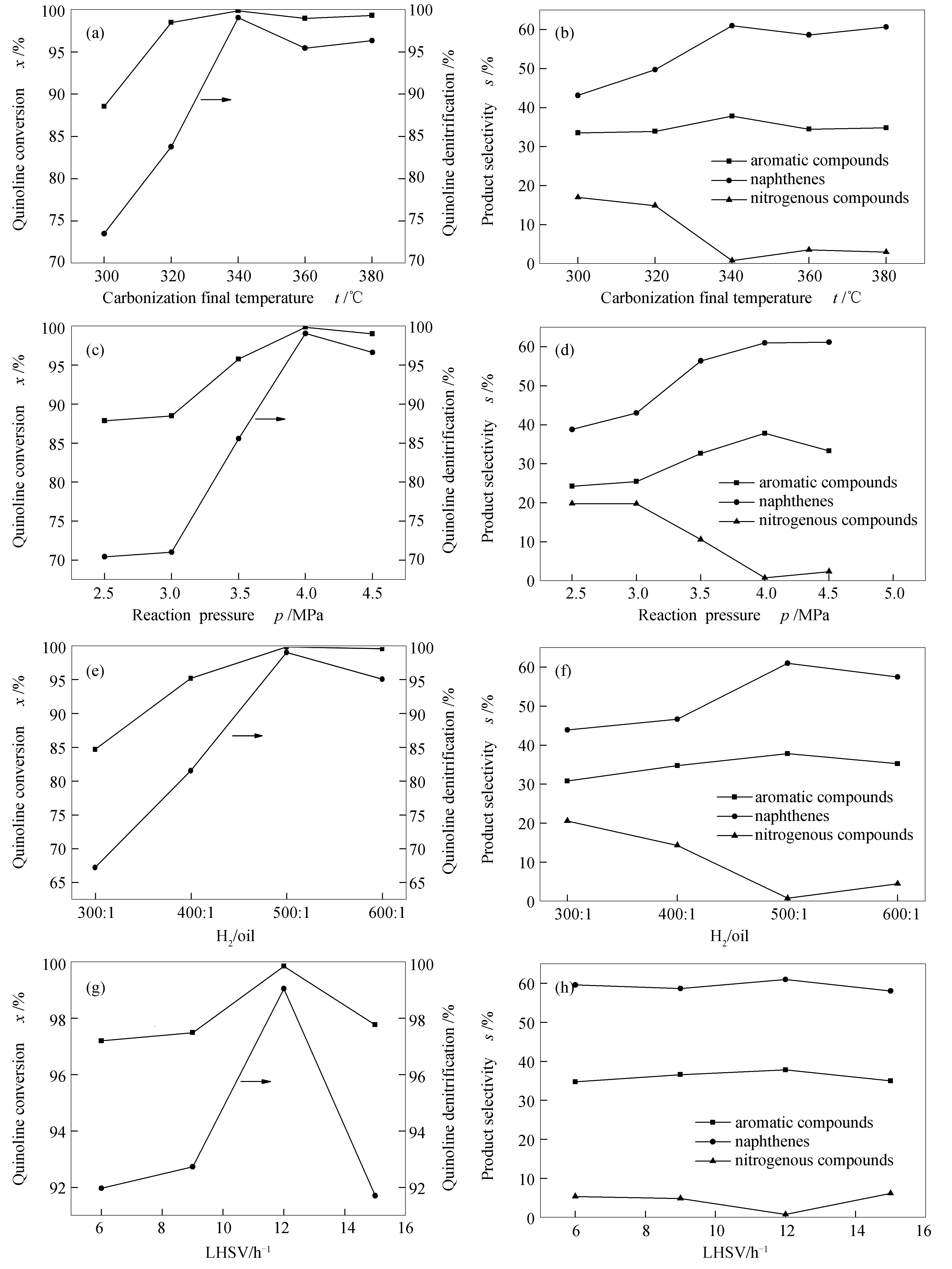
图3 不同反应条件下Mo2C-680催化剂喹啉HDN性能
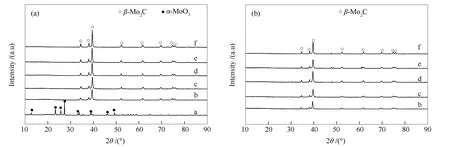
图4 MoO3和不同碳化终温碳化钼催化剂反应前后的XRD谱图
2.2.2 催化剂的N2吸附-脱附表征
不同碳化终温碳化钼的比表面积、孔容以及平均孔径见表1,N2吸附-脱附等温线见图5。由表1可知,随碳化终温升高,碳化钼的比表面积与平均孔径呈现先增大后减小的趋势,但比表面积变化幅度很小。当碳化终温为680 ℃时,碳化钼具有最大的表面积(6.84 m2/g)和平均孔径(47.96 nm),进一步升高碳化终温时,样品的比表面积出现了一定程度的下降,这可能与直接渗碳过程中在表面生成自由碳有关。对比反应前后碳化钼催化剂(碳化终温680 ℃),发现比表面积、孔容以及平均孔径都有一定的减小,结合反应前后并未发生明显的物相破坏(见XRD谱图),这显示在反应过程中可能产生了积炭或团聚现象,这与SEM表征结果一致。由图5可知,样品的吸附-脱附等温线存在明显的回滞环,属于第Ⅳ类吸附等温线,这是因为氮气在介孔内不可逆的吸附和脱附过程导致的。随碳化终温升高,回滞环总体呈现先减小后增加的趋势,碳化终温为680 ℃时,碳化钼回滞环最小,显示出碳化终温影响了碳化钼的介孔分布,其中,680 ℃时碳化钼介孔孔分布相对集中。对比反应前后碳化钼催化剂(碳化终温680 ℃),反应后回滞环明显增大,显示介孔孔分布变得分散,这也与平均孔径的减小相一致。

表1 不同碳化终温碳化钼催化剂的孔结构参数
对于Mo2C-640、Mo2C-660、Mo2C-680、Mo2C-700和Mo2C-720,随碳化终温升高,比表面积、平均孔径的变化趋势与喹啉加氢选择性、活性总变化趋势基本一致,由于各催化剂比表面积相差较小,而平均孔径差别较大,因而平均孔径是影响碳化钼喹啉加氢脱氮性能的重要因素。平均孔径影响到反应场所的大小,理论上必然对反应性能产生一定程度影响,但Mo2C-660和Mo2C-680平均孔径相近(分别为47.40和47.96 nm),加氢脱氮率相差却达到9.0%,芳烃类选择性相差也达到7.0%,Mo2C-680和Mo2C-700平均孔径相差较大(分别为47.96和31.98 nm),加氢脱氮率却仅相差3%,这说明平均孔径并非是影响碳化钼性能的决定性因素。

图5 不同碳化终温碳化钼催化剂的N2吸附-脱附等温线
2.2.3 催化剂的SEM和TEM表征
图6为不同碳化终温碳化钼催化剂的SEM照片(放大10000倍)。由图6可知,Mo2C-640、Mo2C-660、Mo2C-680、Mo2C-700和Mo2C-720的形状均为规整、大小相对均一的片状颗粒,平均粒径约为1 μm。但颗粒形成团聚态,呈现出片状物的堆积体。这可能是因为在碳化过程产生了烧结,从而引起其晶粒的堆积。同时,碳化终温越高((a)-(e)),发生烧结的倾向和程度也略有增大。对比反应前后的碳化钼催化剂(c)与(f)可以发现,其形貌均呈现叶片状的团聚形态,但反应后粒径有所减小,可能是反应过程中颗粒产生了少许的磨损,这可能是导致反应后孔结构改变的原因之一。
图7为MoO3和碳化钼催化剂反应前后的TEM、HRTEM照片。由图7可知,MoO3(图7(a))的形貌为棱柱型、大小不一。碳化钼反应前后(图7(b)、(c))与前驱体MoO3相比,颗粒较为规整且分布均匀,但颗粒堆叠随机形成松散的结构,均有团聚现象发生,且反应后的团聚现象有加重倾向。

图6 不同碳化终温碳化钼催化剂的SEM照片(放大10000倍)
由HRTEM可知,图7(d)中晶格条纹平均间距为0.38 nm,对应于MoO3(110)面的晶格间距。图7(e)和(f)均明显的显示了晶格条纹的平均间距为0.23 nm,与β-Mo2C(200)面的晶格间距保持一致,显示反应前后的样品均为六方相,同时表明该催化剂的整体稳定性较佳,这与XRD结果一致。但反应后碳化钼表面平整度有所下降,显示表面在反应过程中有轻微的磨损,这可能是碳化钼表面元素组成发生变化的原因之一(见表2XPS表征)。
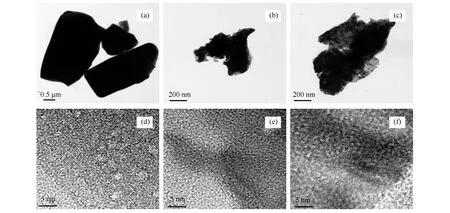
图7 MoO3和碳化钼催化剂反应前后的TEM和HRTEM照片
2.2.4 催化剂的XPS表征
图8为碳化钼催化剂XPS谱图,宽扫描XPS谱图证实了C和Mo元素的存在,同时也检测到O元素的存在(图8(a))。由图8(c)和图8(g)-c可知,不同碳化终温的碳化钼催化剂Mo 3d均在同一结合能处出峰,随着温度的升高,半峰宽、峰强度均略微增大,说明随着温度的升高,碳化程度进一步加深。同时,随碳化终温升高,Mo 3d结合能有逐渐增大的趋势,但增加的幅度很有限(图8(c)),表明Mo物种稳定性提升,这与XRD表征结果一致。Mo 3d的解析光谱均可被分成六个峰,分别对应于Mo6+(233.40和235.95 eV)、Mo4+(229.20和232.30 eV)和Moδ+(228.43和231.90 eV)、(2<δ<4)。其中,Moδ+可归属于Mo-C物种,和碳化钼的形成有关。Mo6+和Mo4+可归因于MoO3和MoO2,这是由于碳化钼材料遇到空气会造成一定的氧化[23]。
随碳化终温升高,C 1s结合能基本保持不变(图8(b)),显示碳化终温并未对C物种结合能产生明显影响。由图8(f)-c可知,C 1s的高分辨XPS谱图可分为三个峰,其中,284.62、285.32和288.87 eV处的碳峰分别归属于C-C、C-H和O-C=O物种[24,25],其分别来源于升温碳化过程中有机气体烃类(CH4)分解产生的碳,直接碳化时有机烃类高温分解脱氢不完全形成的产物,以及碳化钼钝化过程或者暴露在空气与氧形成的产物。
分别对比图8(f)、8(g)中的c和f,可以发现反应后的C-C物种有所增多,这可能是由于在反应过程中形成了一定的积炭引起的。Mo 3d谱图峰强度整体出现了一定的增强,主要源于Moδ+(228.43和231.90 eV)、(2<δ<4)峰的增强,表明更多与C结合的Mo在表面出现,这可能与氢气气流下表面氧的减少相关(见表2)。
表2为不同碳化终温碳化钼催化剂表面原子含量。由表2可知,不同碳化终温制备的催化剂表面原子C、O、Mo的含量都有一定程度的不同,其中,O主要是碳化钼催化剂在O2/N2气流中钝化形成的。随碳化终温升高,C含量先增加后减小,O含量先减小后增大,Mo先减小后增大,Mo2C-680催化剂C含量最高,O含量最低。所有催化剂的C/Mo物质的量比均高于0.5(β-Mo2C理论原子比),说明催化剂表面在碳化过程中会产生积炭,且随着碳化终温的升高,自由碳的含量也有一定程度地增加。同时,随着碳化终温的升高,C/Mo呈现先增加而后减少的趋势,增加主要是因为自由碳的增多,而减小则主要归因于表面Mo含量增加。
对比β-Mo2C(680 ℃)和β-Mo2C(680 ℃-af)可以发现,反应后C/Mo物质的量比较反应前大幅降低,O含量也显著降低,表明有部分表面氧在反应过程中被去除,导致Mo的表面含量有所增大。反应后的碳化钼催化剂表面C/Mo物质的量比仍大于理论原子比,表明反应后催化剂表面仍保持了较好的稳定状态。
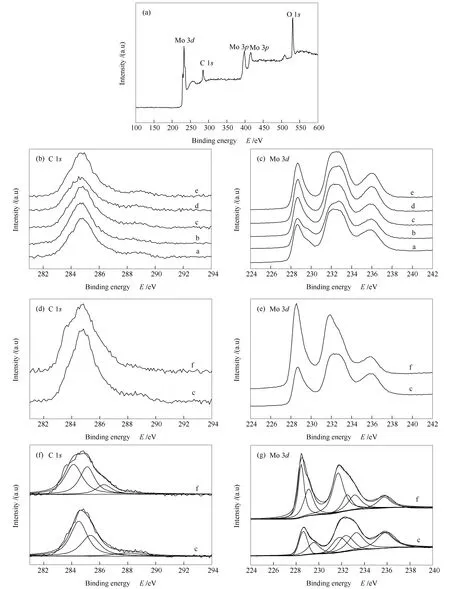
图8 碳化钼催化剂的XPS谱图
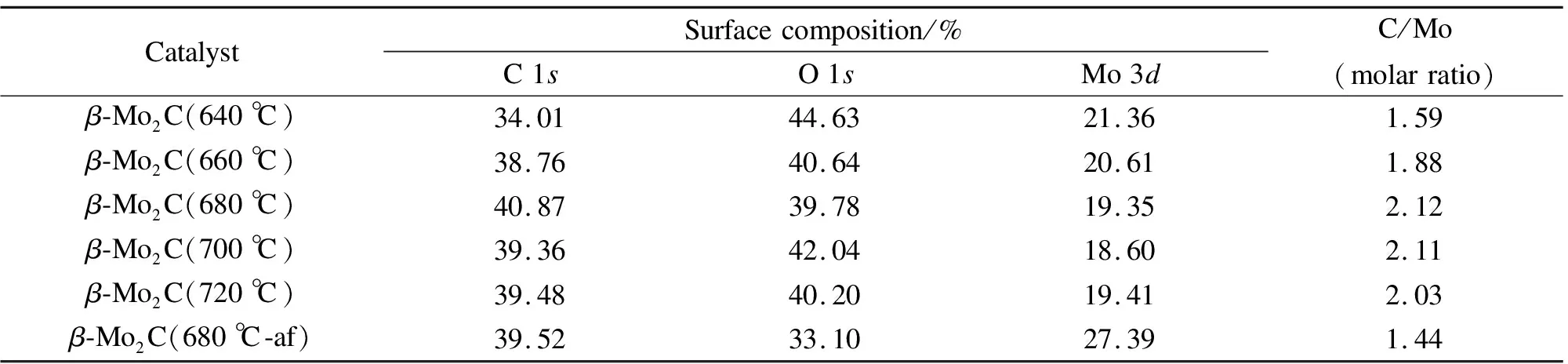
表2 不同碳化终温碳化钼催化剂表面原子含量
2.2.5 催化剂的拉曼表征
图9为MoO3和碳化钼催化剂拉曼光谱谱图(激发波长为532 nm)。

图9 MoO3和碳化钼催化剂拉曼光谱谱图
由图9可知,MoO3(图9a)在995 cm-1(vasMo=O拉伸)、817 cm-1(vasMo-O-Mo拉伸)、665 cm-1(vsMo-O-Mo拉伸)、379 cm-1(Mo=O弯曲)、335 cm-1(Mo=O弯曲)、292 cm-1(Mo=O弯曲)、283 cm-1(Mo=O弯曲)、245 cm-1(Mo=O变形)、218 cm-1(Mo-O-Mo变形)、196 cm-1(Mo-O-Mo变形)、158 cm-1(Mo-O-Mo变形)、127 cm-1(Mo-O-Mo变形)和114 cm-1(Mo-O-Mo变形)处出现拉曼谱带[26],并且819 cm-1处的拉曼谱带峰强度最强。采用20%CH4/80%H2在不同碳化终温下渗碳后(图9b-9f),发现终温为640、660 ℃时,制备的碳化物仍存在995、817 cm-1的拉曼谱带,这是MoO3的特征拉曼散射体,表明表面存在氧化钼。随着温度的升高直至680、700和720 ℃时,拉曼谱带全部减弱,几乎消失,其中,Mo2C-680最弱。鉴于Mo-C对拉曼散射几乎没有作用,结合XRD结果表明,680、700和720 ℃下的碳化产物是纯度极高的β-Mo2C(HCP)。
图9g显示了碳化终温为680 ℃的反应后碳化钼催化剂的拉曼光谱谱图(激发波长为532 nm)。对比图9d与9g,发现经过反应后,在995、817、292和218 cm-1等处有很弱的拉曼谱带出现,显示表面检测到了氧化钼,由于反应系统内无外来氧,而同时反应后Mo 3d峰增强(图8(g)XPS谱图),可推断,氧化钼出现是由反应中催化剂表面状态发生变化所致。显然,表面氧化钼越多,越不利于碳化钼喹啉加氢脱氮反应(图2)。
2.3 碳化钼性质改变对其喹啉HDN性能的影响
前文表明,碳化终温变化未改变催化剂体相的物相,在较小程度上影响结晶度、物相稳定性、比表面积、催化剂形貌、C和Mo物种结合能等,但可以显著改变碳化钼平均孔径和表面物种含量。各碳化终温制备的碳化钼体相均为β-Mo2C,评价数据(图2)表明,β-Mo2C显然是喹啉加氢脱氮的活性相,但体相不是造成碳化钼喹啉加氢脱氮性能差别的原因,平均孔径也并非是决定性因素。因此,在反应条件被限定时,对催化剂活性位有显著影响的催化剂表面状态和组成应是关键影响因素。XPS和Raman显示,催化剂表面物种和元素组成较为复杂。不同碳化终温催化剂表面物种种类相同,结合能也相近,但由表面C、Mo和O含量来看,表面物种的含量有明显差异。Mo2C-680表面氧含量最低,氧化钼最少,具有最佳的喹啉加氢脱氮活性和芳烃类产物选择性。同时,不同碳化终温催化剂氧含量呈现先减小后增加的趋势,这与各催化剂喹啉加氢脱氮活性和芳烃类产物选择性变化趋势相反,可知表面氧的量与碳化钼活性和芳香烃选择性密切相关,但对环烷烃类选择性影响较小。Lee等[27]使用化学滴定和瞬态反应动力学研究了表面氧对Mo2C苯甲醚加氢脱氧反应的影响,结果显示表面氧的增加会减少加氢脱氧活性位的数量。Schaidle等[28]以乙酸加氢反应研究了碳化钼表面化学和活性位,结果表明,碳化钼表面存在类金属的氢吸附位(暴露在表面的Mo和C)和氧空位(暴露在表面的Mo)等活性位,表面氧对活性位有显著影响。尽管反应底物不同,上述文献也可在一定程度上佐证本研究关于表面氧的结果,可以推断,对于喹啉加氢脱氮反应,碳化钼上可能存在不止一种活性位,而表面氧的增加可对促进芳香烃生成的活性位产生不利影响。
综上,β-Mo2C具有较高的喹啉加氢脱氮活性和芳烃类产物选择性,碳化终温可显著影响碳化钼表面的物种含量,其表面组成可影响活性位进而影响喹啉的加氢脱氮活性和选择性,其中,氧含量的影响最为显著,因而碳化钼催化剂上喹啉的加氢脱氮是表面敏感反应,这也提示,表面组成尤其是表面氧对于调控β-Mo2C上喹啉加氢脱氮反应途径至关重要。
3 结 论
不同碳化终温(640、660、680、700和720 ℃)下均获得了较高纯度的β-Mo2C,碳化钼平均粒径约为1 μm。碳化终温可以显著改变碳化钼表面物种含量、平均孔径和介孔分布。碳化终温提高有利于Mo物种的稳定,但会产生轻微的烧结倾向。碳化过程中在催化剂表面形成自由碳,碳化钼表面C/Mo物质的量比明显高于理论原子比。碳化温度为680 ℃时β-Mo2C表面氧物种含量最低,平均孔径最大,C/Mo物质的量比最高。反应后的碳化钼催化剂未检测到新的物相,但比表面积和平均孔径减小,孔径分布变得分散,C/Mo物质的量比显著下降。
β-Mo2C上喹啉的加氢脱氮是表面敏感反应,表面组成是影响其喹啉加氢脱氮活性和选择性的关键因素,其中,表面氧的影响最为显著。表面组成尤其是表面氧对于调控β-Mo2C上喹啉加氢脱氮反应途径至关重要。碳化终温为680 ℃的β-Mo2C具有最佳的催化活性,喹啉转化率和脱氮率均可达99%,同时对芳环类产物的选择性可以达到37.8%。反应压力对产物选择性影响显著。β-Mo2C具有一定的使芳环侧链C-C键断裂的性能。
猜你喜欢
中国防痨杂志(2022年7期)2022-11-25
四川水泥(2022年10期)2022-11-17
农业工程学报(2022年10期)2022-08-22
钢铁钒钛(2022年2期)2022-08-03
煤炭工程(2022年7期)2022-07-21
工业建筑(2022年2期)2022-06-29
炭素(2021年3期)2021-12-31
纺织检测与标准(2021年1期)2021-12-05
绵阳师范学院学报(2021年11期)2021-11-24
科技与创新(2020年16期)2020-08-18
