
取代水杨醛席夫碱金属配合物结构对硫化物催化氧化性能的影响
2020-05-27 01:13姜翠玉梁书源贾超洋张龙力
石油学报(石油加工) 2020年3期
姜翠玉, 梁书源, 贾超洋, 刘 蕾, 邵 雪, 张龙力
(1.中国石油大学(华东) 理学院,山东 青岛 266580;2.中国石油大学(华东) 化学工程学院,山东 青岛 266580)
硫和含硫化合物广泛存在于石油产品中,如何降低油品的硫含量是储运、加工和使用这些油品时面临的一项巨大挑战。燃料油脱硫方式主要有碱洗脱硫[1]、加氢脱硫[2-4]和氧化脱硫[5-7]。传统的碱洗脱硫方法仅能脱除部分酸性硫化物,如硫化氢和小分子硫醇等,但会产生大量有害硫酸盐废液和废碱液,易造成环境污染;加氢脱硫方法尽管脱硫效率较高,但很难脱除杂环硫化物,反应条件苛刻,且成本高、投资大。相比之下,氧化脱硫的脱硫效率较高、条件更温和[8]、成本更低、适用性更强。但目前的氧化脱硫工艺中,氧化剂常用过氧化氢和有机过氧化物,消耗量巨大且存在严重的安全隐患[9]。
席夫碱金属配合物是一种高效的催化氧化反应催化剂[10-11],已被用于苯甲醇[12]、环己烷[13-14]等化合物的催化氧化,但迄今未见用于油品氧化脱硫的研究报道。如果席夫碱金属配合物对油品中的硫化物的脱除具有明显的催化效果,则可能发展成为一个新的研究领域,并有可能影响传统的脱硫技术。
笔者以具有不同电子效应和位阻效应取代基的水杨醛为原料,分别与乙二胺和邻苯二胺反应合成7种席夫碱配体,然后将合成的配体分别与硝酸钴和硝酸镍反应,制备7种席夫碱金属配合物(C1~C7),并考察了其摩尔载氧量与溶解度。然后以合成的配合物作为催化剂,以氧气为氧化剂,考察其对含1-己硫醇、二丁基硫醚和2-甲基噻吩的模型硫化物体系的催化氧化性能,探讨影响席夫碱金属配合物催化氧化脱硫活性的因素,并从中心金属离子种类、配体的电子效应和空间效应以及配合物溶解度等方面进行配合物构效关系探究,为进一步开展席夫碱金属配合物在燃料油催化氧化脱硫方面的研究和应用打下一定的基础。
1 实验部分
1.1 试剂与仪器
试剂:乙醇、硝酸钴、硝酸镍、正辛烷、氢氧化钠,均为分析纯,国药集团化学试剂有限公司产品;乙二胺、邻苯二胺、水杨醛、邻香兰素、5-氯水杨醛、3,5-二叔丁基水杨醛、1-己硫醇、二丁基硫醚、2-甲基噻吩、N,N-二甲基甲酰胺,均为分析纯,阿拉丁试剂公司产品。
仪器:RE-52A型旋转蒸发仪,上海亚荣生化仪器厂产品;7820A型气相色谱,安捷伦(上海)科技有限公司产品。
1.2 席夫碱金属配合物的合成
按照反应式(1)所示,分别将水杨醛、邻香兰素、5-氯水杨醛、3,5-二叔丁基水杨醛与乙二胺按n(醛)∶n(胺)=2∶1比例制备席夫碱配体L1~L4;按照反应式(2)所示,将水杨醛、3,5-二叔丁基水杨醛与邻苯二胺按n(醛)∶n(胺)=2∶1比例制备席夫碱配体L5和L6。然后,将L1按照反应式(3)分别与Co(NO3)2、Ni(NO3)2反应合成配合物C1和C2;将L2~L4按反应式(3)与Co(NO3)2反应合成配合物C3~C5;将L5、L6按式(4)与Co(NO3)2反应合成配合物C6、C7。具体合成步骤参照文献[15]进行。

(1)
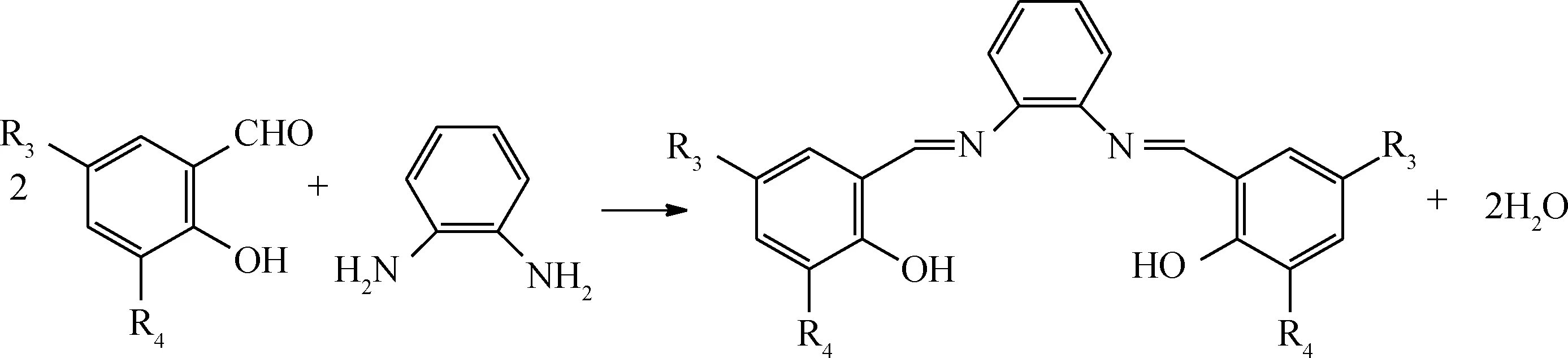
(2)
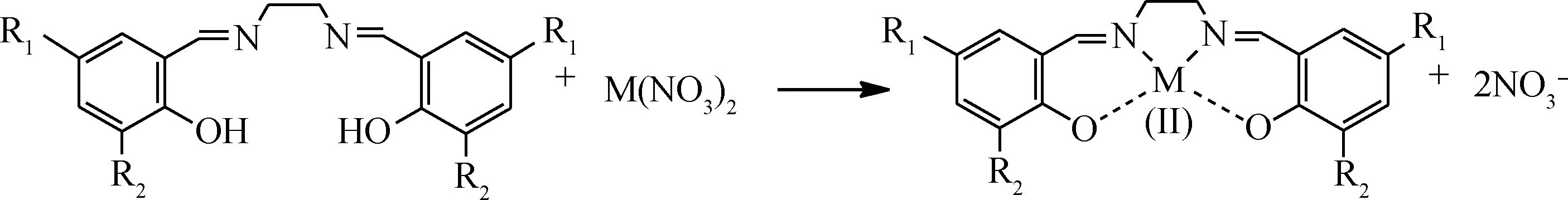
(3)
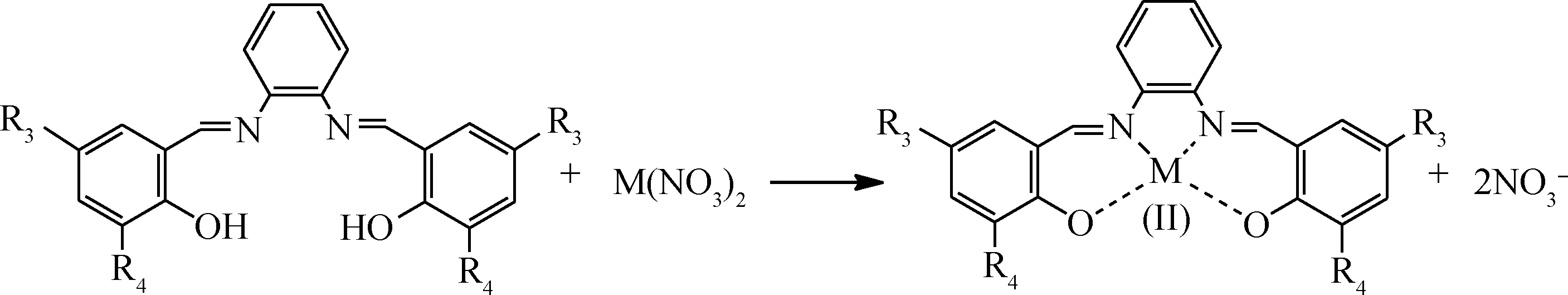
(4)
1.3 配合物载氧量及溶解度测定
1.3.1 配合物载氧量测定
根据文献[16]设计载氧实验装置,如图1所示。以C1为例,称取0.3 g配合物C1置于圆底烧瓶中,在恒压漏斗中加入30.0 mL的DMF(N,N-二甲基甲酰胺)作溶剂;向量气管中注入一定量的水,记录水面初始高度,保证量气管垂直;向实验体系通入O2,检验并确保实验装置的密封性。记录室温T以及大气压p。将DMF加入烧瓶,开启搅拌并计时。每隔2 min记录量气管液面变化,直至液面恒定。绘制载氧体积V随时间t变化的曲线,得到0.3 g配合物最大载氧量后,由式(5)计算出每摩尔配合物吸收的O2的物质的量。
(5)
式(5)中,n为摩尔载氧量,即每摩尔配合物吸收O2的物质的量,mol/mol;m为配合物质量,g;M为配合物分子相对分子质量,g/mol;p为实验进行时的大气压,Pa;V为吸收O2的体积,mL;R为理想气体常数,J/(mol·K);T为实验进行时的室温,K。
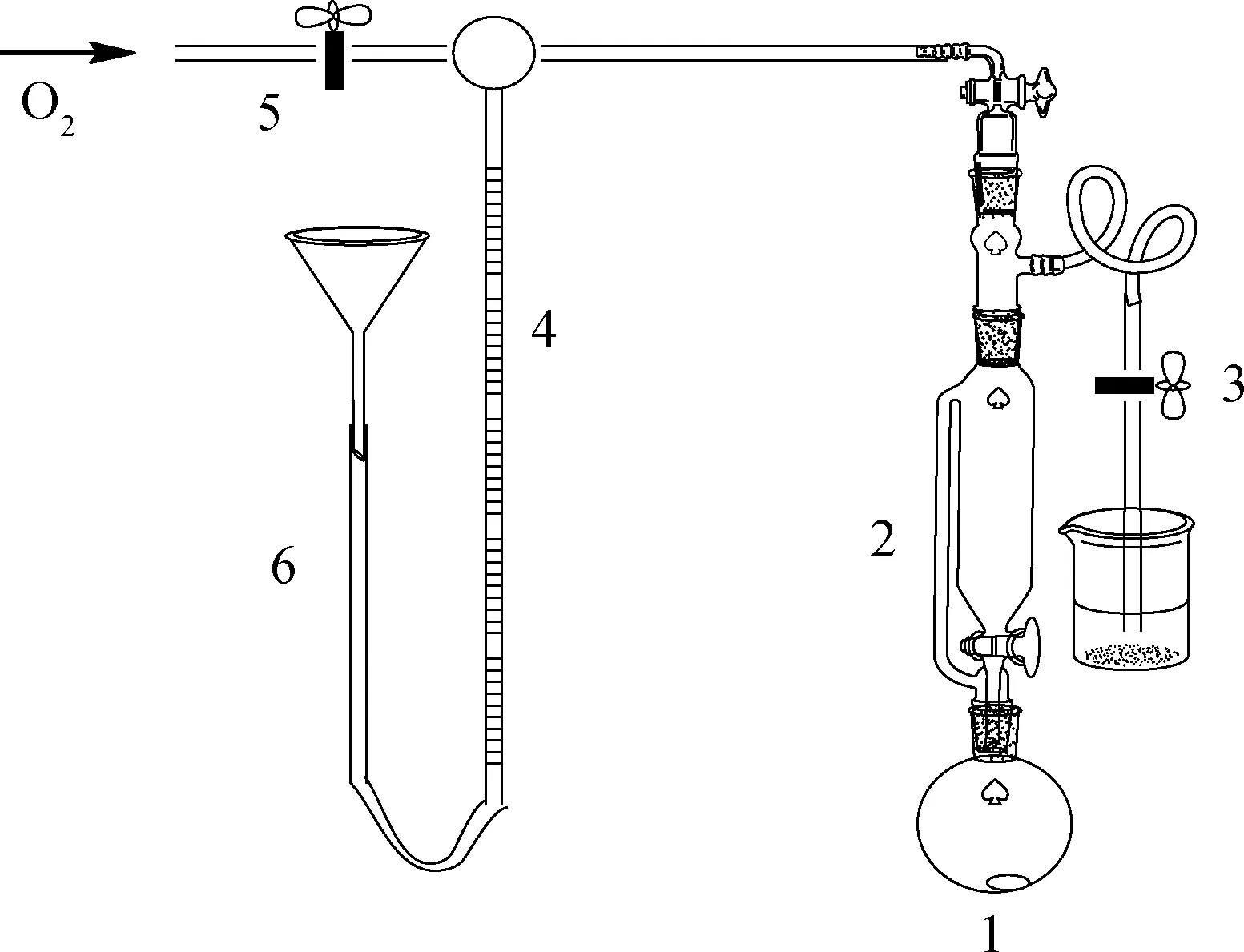
图1 载氧实验装置Fig.1 Oxygen carrying experimental device1—Round-bottomed flask; 2—Constant pressure funnel;3,5—Cock; 4—Trachea; 6—Glass tube
1.3.2 配合物在DMF及正辛烷中溶解度测定
由于DMF是载氧实验所用溶剂,而正辛烷是模拟油体系的溶剂,配合物在二者中的溶解度会影响载氧性能和反应性能。故采用静态平衡法[17]测定C1~C7在DMF和正辛烷中的溶解度。方法:将配合物分别溶解于DMF和正辛烷,得到饱和溶液;称量空圆底烧瓶质量m2,取10.0 mL配合物饱和溶液于烧瓶中,蒸干溶剂,烘干至恒重,称重得m1,配合物溶解度S根据式(6)计算。
S=(m1-m2)/10
(6)
式(6)中,m1为蒸干溶剂后剩余溶质和圆底烧瓶的质量,g;m2为圆底烧瓶质量,g;S为配合物溶解度,g/mL。
1.4 模型硫化物的催化氧化性能考察
1.4.1 模拟油催化氧化实验
将1.0 mL 1-己硫醇、二丁基硫醚、2-甲基噻吩分别加入 200.0 mL 正辛烷中,配制成模拟油。在带有磁力搅拌、冷凝管的250 mL三口烧瓶中依次加入 0.10 g 配合物、100.0 mL模拟油和30 mL的NaOH水溶液(质量分数为30.0%)。水浴温度调节至80 ℃,搅拌转速1200 r/min,使得水溶液与模拟油层充分混合,以0.80 L/min的气速向体系中通入氧气进行反应。考察氧化时间为15、30、45、60、75 min时,配合物对模拟油中硫化物的催化氧化效果。
1.4.2 模型硫化物氧化转化率测定
1-己硫醇、二丁基硫醚、2-甲基噻吩在配合物C1~C7催化下,被氧气氧化后皆生成相应的砜类或亚砜类物质。其中1-己硫醇、二丁基硫醚还进一步被氧化生成SO32-或SO42-。按照文献[18]采用气相色谱分析氧化前、后体系中3种模型硫化物的含量,计算其氧化转化率。分析前,在油样中加入1.0 μg的1-己硫醇、二丁基硫醚和2-甲基噻吩标准物质,根据标准物加入前、后峰面积的变化,确定各硫化物含量。气相色谱分析条件为:HP-5色谱柱(30.0 m×0.25 mm×0.25 μm);FID检测器温度为300 ℃;采用程序升温法升温;分流比为10∶1;载气为氮气,流量为20 mL/min。
为了避免硫化物的挥发引起的测量误差,氧化t分钟后油样中硫化物a的质量mat计算式如式(7)所示。
(7)
式(7)中,A和A1分别为标准物质加入前和加入后硫化物a的峰面积。氧化t分钟后硫化物a的氧化转化率ηat计算公式如式(8)所示。
(8)
式(8)中,ma和mat分别为氧化前、后油样中硫化物a的质量,mg。
2 结果与讨论
2.1 配合物的载氧性能及溶解度
2.1.1 配合物的载氧性能
7种配合物的摩尔载氧量和达到最大载氧量的时间如表1所示,其中n为摩尔载氧量,tmax为达到最大载氧量所需要的时间。由表1可知,7种配合物的摩尔载氧量由高到低顺序为C7、C6、C3、C1、C2、C5、C4,每摩尔C7配合物最多可以吸收0.523 mol O2。席夫碱配合物的载氧性能主要由其结构决定:中心离子与氧分子的配位能力越强、配体取代基推电子能力越大且空间位阻越小、共轭链越长,越有利于载氧。如:由邻苯二胺制得的配合物C7、C6的载氧能力明显高于以乙二胺合成的配合物C1~C5,因为邻苯二胺分子的共轭程度更大,配合物与O2结合形成的中间体结构更稳定,其载氧能力也更好;而C1与C2相比,席夫碱配体相同,中心金属离子分别为Co(Ⅱ)和Ni(Ⅱ)离子,Co(Ⅱ)离子的电子密度高于Ni(Ⅱ)离子,因此C1吸附氧的能力优于C2,载氧能力更强;对于C3、C1、C5和C4,其载氧能力因配体苯环上的取代基的不同而不同。C3中苯环3号位含有强推电子基团-OCH3,使苯环电子云密度增大并通过共轭效应增强了中心金属离子的电子云密度,因此其载氧能力最大。C4苯环含吸电子基团-Cl,降低了中心金属离子的电子云密度,因此其载氧性能较差。C5尽管含有推电子基团-C(CH3)3,但由于2个叔丁基尤其是3号位的叔丁基存在较大的空间位阻效应,影响配合物的共平面性从而影响其共轭的程度,造成其载氧性能较低。
此外,7种配合物达到最大载氧量的速率由大至小顺序为C4、C5(C3)、C6、C1、C7、C2。其中,C5与C3载氧速率相同。

表1 C1~C7摩尔载氧量与达到最大载氧量的时间Table 1 Molar oxygen carrying property and the time to reach maximum oxygen carrying of C1-C7
2.1.2 配合物在不同溶剂中的溶解度
室温下,配合物C1~C7在DMF和正辛烷中的溶解度如表2所示。由表2可知,C1~C7在DMF中都能较好地溶解;而在正辛烷中溶解度较小且差别较大。其在正辛烷中的溶解度由大到小顺序是C5、C6、C7、C2、C1、C4、C3。分析原因,C5苯环上带有2个疏水性-C(CH3)3基团,极性最小,在正辛烷中的溶解度最高;C6和C7中含有更多的疏水性芳香环,溶解度也较高;而C3因苯环上的-OCH3有一定亲水性,使其在正辛烷中的溶解度最低。在正辛烷中溶解度越大,意味着配合物在溶剂中分散得越均匀,有效浓度越高,氧化过程中的传质效果更好。配合物不溶解的部分是以固体颗粒的形式漂浮于油-水界面或者沉降至底部,对氧化过程中的传质作用有较大的不利影响。
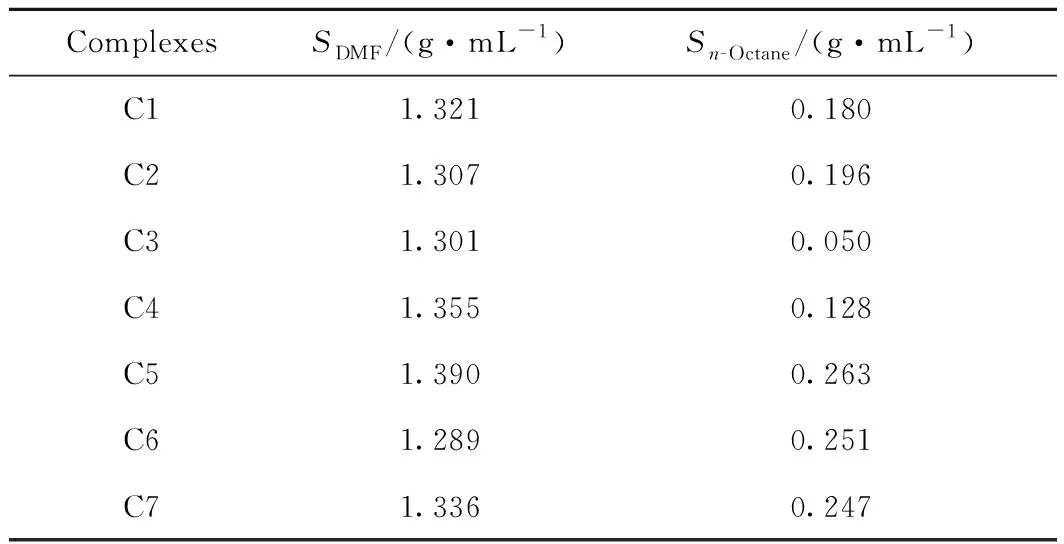
表2 配合物C1~C7在DMF以及正辛烷中的溶解度Table 2 The solubility of C1-C7 in DMF/ n-octane
2.2 配合物的催化氧化性能考察
对模拟油进行催化氧化实验发现,由于NaOH水溶液对硫醇具有碱洗脱除作用,1-己硫醇在 10 min 左右就接近100%的转化率[15]。为排除NaOH水溶液碱洗作用对硫醇等脱除率的影响,催化氧化反应在非碱性体系中进行。反应75 min内,模拟油中1-己硫醇、二丁基硫醚和2-甲基噻吩的氧化转化率随氧化时间的变化曲线如图2所示。其中,图2(a)~(g)分别对应配合物C1~C7催化的氧化脱硫反应。
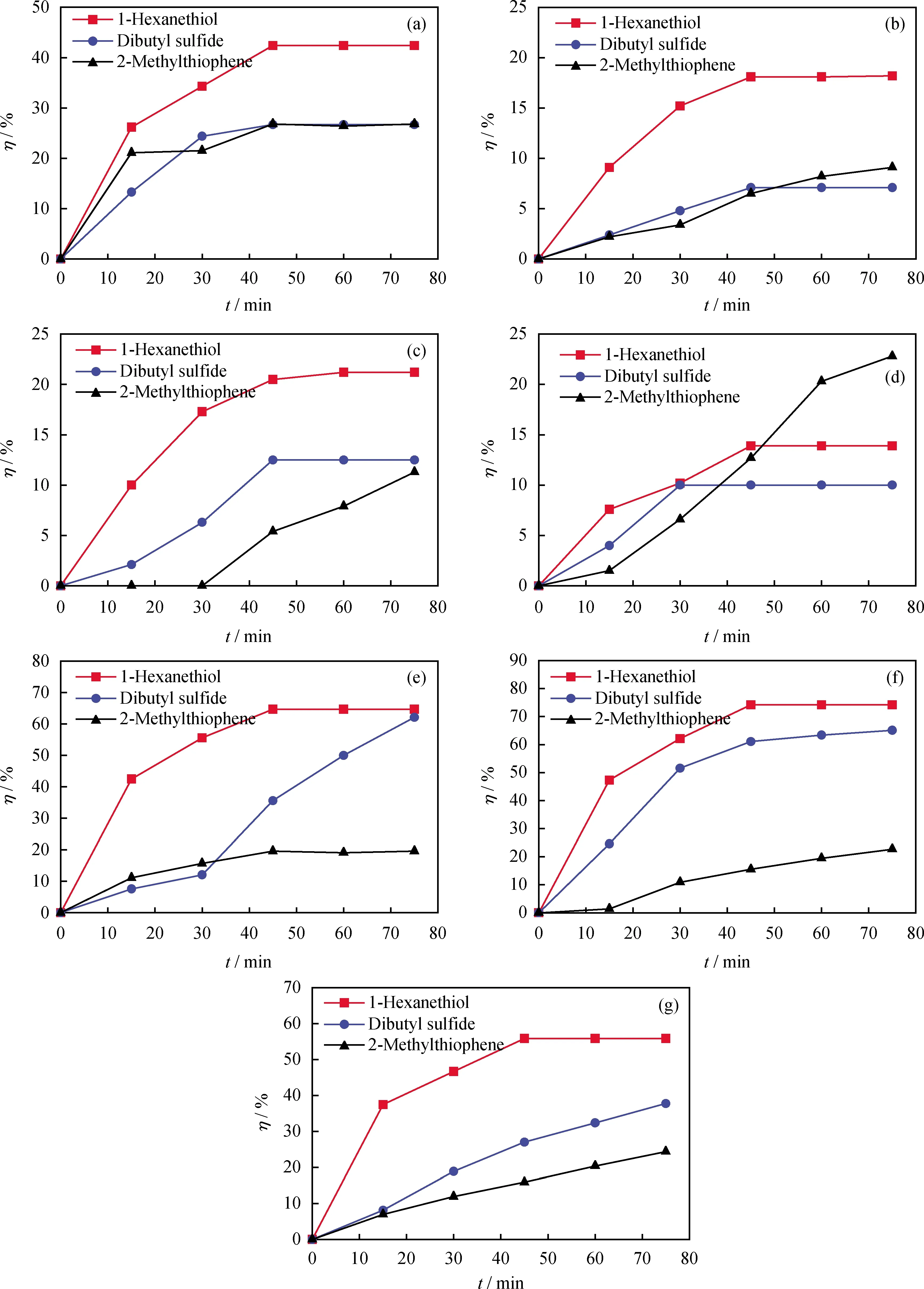
图2 不同配合物催化下各硫化物氧化转化率与时间的关系Fig.2 Relationship between conversion rate and time of various sulfides catalyzed by different complexes(a) C1; (b) C2; (c) C3; (d) C4; (e) C5; (f) C6; (g) C7Reaction conditions: m(Catalyst)=0.1 g; F(O2)=0.8 L/min; V(Oil)=100.0 mL
由图2可知:随着反应时间的延长,3种硫化物在不同配合物催化下的氧化转化率基本上都呈先上升后平稳的趋势;C4(图2(d))对三者的氧化性能都较弱;除以C4为催化剂的反应外,其他催化氧化反应中1-己硫醇的氧化转化率均高于二丁基硫醚和2-甲基噻吩;C5(图2(e))、C6(图2(f))催化二丁基硫醚的氧化转化率明显高于2-甲基噻吩,趋近于1-己硫醇;配合物C6对3种硫化物的催化氧化转化率都较高,1-己硫醇的氧化转化率最高可达74.2%,二丁基硫醚的氧化转化率为65.1%,而7种配合物对2-甲基噻吩的催化氧化效果都较差,主要是因为2-甲基噻吩环系较稳定,且噻吩硫上的电子云密度较低[19],不易被氧化。
2.3 影响配合物催化氧化性能的因素
2.3.1 中心金属离子对配合物催化性能的影响
配体相同,中心金属离子分别为Co(Ⅱ)和Ni(Ⅱ)离子的配合物C1和C2催化氧化3种硫化物的转化率比较如图3所示。由图3可知,C1对3种硫化物的催化氧化性能均优于C2。原因在于,C1中心金属离子Co(Ⅱ)更易与氧气形成配合物,使C1吸附、活化分子氧的能力和稳定性都好于C2[13],即C1载氧性能优于C2。同时,二者在正辛烷中的溶解度相近,说明其催化氧化性能主要受其载氧性能影响。因此,配体相同情况下,中心金属离子为Co(Ⅱ)的配合物的催化性能优于中心离子为Ni(Ⅱ)的配合物。
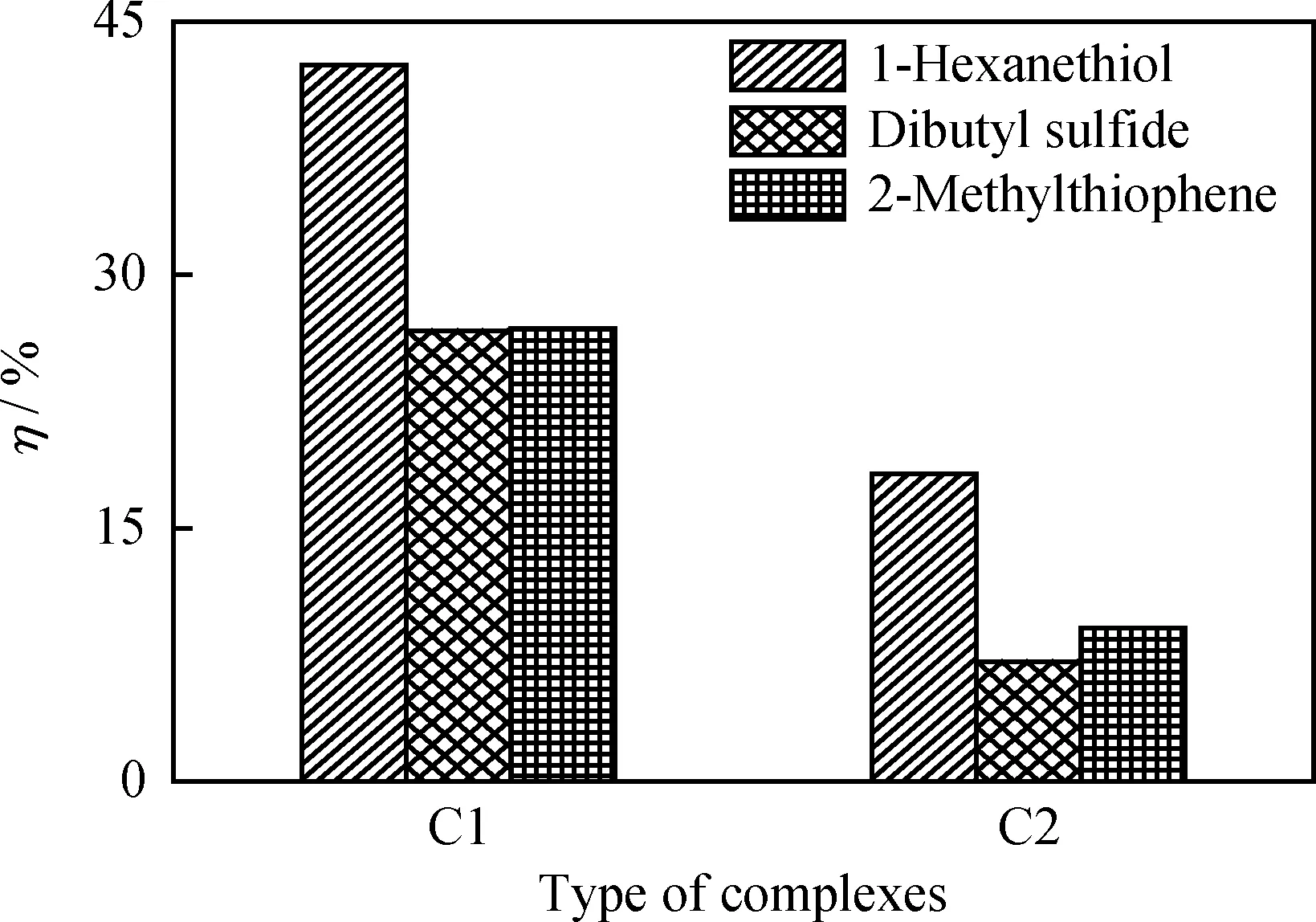
图3 中心离子对配合物催化氧化性能的影响Fig.3 Effects of the central ion on catalytic oxidation property of complexesReaction conditions: m(Catalyst)=0.1 g; F(O2)=0.8 L/min;V(Oil)=100.0 mL; t=75 min
2.3.2 水杨醛苯环上的取代基对配合物催化性能的影响
考察C1、C3、C4、C5对模型硫化物的催化氧化性能,结果如图4所示。由图4可知,配合物催化氧化硫化物性能由高到低的顺序为C5、C1、C3、C4。配合物的催化氧化性能与其载氧性能规律并不完全一致,说明还可能受其他因素影响。
配合物C1、C3、C4、C5结构上的主要区别在于水杨醛苯环上取代基的不同:C1中水杨醛苯环上没有取代基;C3、C5苯环上分别有推电子基 -OCH3和-C(CH3)3,且推电子能力-OCH3大于-C(CH3)3;而C4苯环上连有吸电子基-Cl。图4 结果表明,苯环上有吸电子基的配合物对硫化物的催化氧化性能最差;苯环上有推电子基的配合物性能较好;但含有强的推电子基-OCH3的配合物C3的催化氧化性能却低于没有取代基的C1,这说明配合物的催化氧化性能不仅仅取决于取代基的吸、推电子能力,还受其在溶剂中溶解性能的影响。尽管-OCH3的推电子能力最强,使C3的载氧量最大,但是由于其亲水性较强,导致C3在正辛烷中的溶解度最小,从而影响C3对模拟体系中硫化物的催化氧化性能。
因此,配合物对硫化物的催化氧化性能受水杨醛苯环上取代基及其在体系中的溶解度的共同影响。在选择原料水杨醛时,既要考虑取代基的电子效应又要考虑其溶解性。C5的载氧能力虽然稍差,但是其在正辛烷中的溶解度最大,较高的浓度有利于配合物与硫化物的相互作用,弥补了其载氧性能不佳的劣势,使其具有相对更好的催化氧化性能。
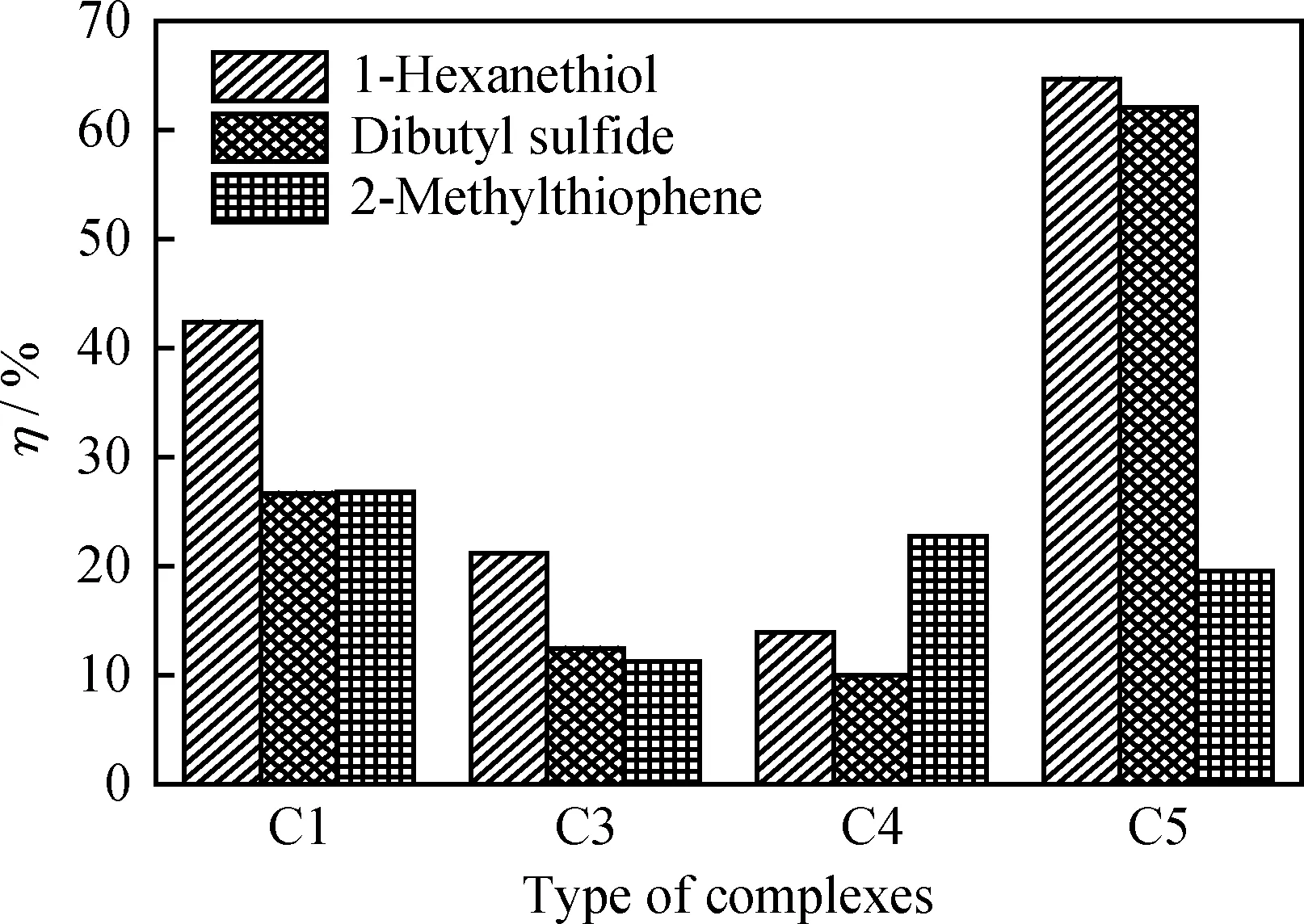
图4 苯环取代基对配合物催化氧化性能的影响Fig.4 Effects of substituent groups on catalytic oxidation property of complexesReaction conditions: m(Catalyst)=0.1 g; F(O2)=0.8 L/min;V(Oil)=100.0 mL; t=75 min
2.3.3 反应物胺的结构对配合物催化性能的影响
配合物C1和C6对硫化物的催化氧化性能如 图5 所示。由图5可知,C6的催化氧化性能比C1更好。分析原因主要是二者合成原料二胺的结构不同所致,C6是由邻苯二胺制备得到的,其分子的整体共轭程度大于由乙二胺合成得到的C1,配合物C6与O2结合形成的中间体结构更稳定,使其载氧性能和在正辛烷中的溶解度都比C1更大。
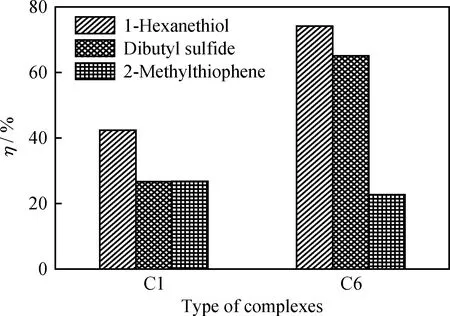
图5 不同的二胺对配合物催化氧化性能的影响Fig.5 Effects of different diamines on catalytic oxidation property of complexesReaction conditions: m(Catalyst)=0.1 g; F(O2)=0.8 L/min;V(Oil)=100.0 mL; t=75 min
3 结 论
(1)配合物C1~C7在DMF中溶解度较大且几乎没有差别;在正辛烷中溶解度差别较大,且由大到小顺序是:C5、C6、C7、C2、C1、C4、C3;配合物中含有的疏水性基团越多,其在非极性溶剂正辛烷中的溶解度则越大。
(2)配合物C1~C7的载氧能力由高到低的顺序为C7、C6、C3、C1、C2、C5、C4。配合物中心金属离子电子云密度越大,配体取代基推电子能力越大、空间位阻越小、共轭链越长,则其载氧能力越强。
(3)配合物C1~C7催化氧化硫化物的性能受其载氧能力和在正辛烷中的溶解度共同影响,其载氧能力越强,在正辛烷中溶解度越大,则催化氧化硫化物的性能越强。配合物对不同硫化物的催化氧化能力不同。对3种硫化物催化氧化性能效果最好的配合物为C6,其催化氧化1-己硫醇的转化率达到74.2%,二丁基硫醚的转化率为65.1%,2-甲基噻吩的转化率较低。
猜你喜欢
能源化工(2022年1期)2023-01-14
汽车实用技术(2022年7期)2022-04-20
太原理工大学学报(2022年2期)2022-03-21
湖北农机化(2021年7期)2021-12-07
化工环保(2021年3期)2021-06-17
石油学报(石油加工)(2021年1期)2021-01-27
石油石化绿色低碳(2019年6期)2019-01-14
中国塑料(2016年8期)2016-06-27
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
中学科技(2015年8期)2015-08-08
