
基于全基因组重测序技术分析甜菜InDel标记
2020-07-16 04:36黄平仙高永明刘乃新付畅吴玉梅
中国糖料 2020年3期
黄平仙,高永明,刘乃新,付畅,吴玉梅
(1.黑龙江大学现代农业与生态环境学院,哈尔滨 150080;2.哈尔滨师范大学生命科学与技术学院,哈尔滨 150025)
0 引言
甜菜属于石竹目、藜科、甜菜属,甜菜最初生长于欧洲西南部地区,是甘蔗以外的第二大糖料来源。甜菜产业在我国北方占有重要地位,甜菜糖业在西北、东北稳定发展,在华北呈不断提升发展态势[1]。分子标记、基因工程、组学技术等为甜菜的深入研究提供了重要技术手段[2]。在遗传育种研究中能够快速、高效且准确地识别甜菜品种十分重要。目前应用于物种鉴定试验中的分子标记技术主要有SCoT[3]、SSR[4]和SRAP[5]等,主要用于甜菜物种鉴定的技术有SSR[6]、SRAP[7]和ISSR[8],每种方法各有长处。
全基因组重测序技术是针对人类已经掌握全部基因组序列的个体进行基因组测序,并将所取得的结果与个体、群体的基因组进行差异性分析,将测序后得到的结果与已经掌握的基因组进行比对,可以检测出全基因组内的插入、缺失突变和核苷酸序列改变等多种变异信息,全基因组重测序后得到的数据是鉴定经济动、植物遗传性状的重要依据,可根据基因组变化的结果针对多种突变进行开发利用,为各种经济作物的品种筛选提供便利[9]。InDel 标记是根据不同个体基因组同源序列发生的核苷酸片段插入或缺失而开发的,InDel位点在基因组内分布广、密度高、变异率高,InDel标记技术的可重复性好并且成本不高昂[10-11]。因此利用全基因组重测序技术开发InDel 标记,并利用这种方法进行品种鉴定已经被广泛应用于水稻[12]、朝天椒[13]和番茄[14]等农作物中,诸多研究已经证明了InDel标记技术的可靠性,因此将其应用于甜菜品种的筛选是可行的方案。
2016年,Ries等利用不同表型育种系品种通过深度测序和InDel标记法对甜菜不同基因型样本进行了快速检测[15]。但是,目前已知的甜菜InDel标记较少,研究相对落后,将InDel标记应用于甜菜品种的筛选可以从多种角度、更加全面地筛选不同品种的甜菜。本研究利用全基因组重测序技术对5 个甜菜品种的基因组进行了测序,第二代测序技术能够高效、快速和准确地获得5 种甜菜全基因组序列,进而可将其基因组上的插入与缺失位点开发出一套全新的InDel引物,为甜菜种质资源的筛选提供更加多样化、细致化的选择基础,对甜菜品种的保护起到重要的作用。
1 材料和方法
1.1 试验材料
以‘MA3001’、‘KWS1231’、‘ZT000589’、‘ZT000549’、‘ZT000286’五个甜菜种质资源为试验材料,从每个甜菜品种中取出15 粒种子,种植在大小适宜的花盆内,待植株的茎长到5~6 cm后将茎剪下来,快速地封装在做好标记的容器中,而后转移到超低温冰箱中留用。
1.2 试验方法
1.2.1 甜菜基因组DNA的提取
采用CTAB法提取甜菜基因组的DNA,并用1%琼脂糖凝胶电泳对DNA样品进行质量检测[16]。
1.2.2 文库构建与测序
使用Covaris破碎机将检验后无污染且纯度好的DNA 样品随机切断成长度约为350 bp 的DNA 片段,而后使用Klenow exo 对DNA 片段进行前端修复、加尾和连接测序接头,最后使用TruSeq Library Construction Kit回收DNA片段,得到甜菜的基因组测序文库。
构建测序文库后,用核酸蛋白定量仪Qubit2.0 进行初阶定量,将基因组浓度减少到1 ng/µL,然后使用Agilent 2100 针对文库的插入尺寸进行检测,符合预期后对文库的有效DNA 浓度进行定量(文库浓度应>2 mol/L)。在保证文库质量的基础上进行illumina HiSeq测序。
1.2.3 测序质量检查
按表1中的标准进行测序错误率检测,测序错误率用e表示,碱基质量值Qphred=-10 log10(e)。

表1 测序正确率与Phred 分值的对应关系Table 1 The correlation between sequencing accuracy and Phred score
1.2.4 测序数据过滤
测序后得到的序列是原始数据,中间掺杂有接头的低品质的数据。为确保测定数据的品质,需要对原始数据进行过滤,将有接头的数据去掉,将单头测序中含有N 比例超过10%的数据去掉,将单头测序中低质量碱基大于50%的数据去掉,得到有效数据,后续分析都根据有效数据展开。
1.2.5 重测序序列与参考基因组的比对及InDel位点的检测
对重测序数据进行过滤后,使用BWA 软件[17]将有效数据与参考基因组的序列进行比对,寻找一致的基因组数据,然后使用SAMTOOLs软件[18]检测一致基因组数据中的插入及缺失位点。
1.2.6 InDel位点的分布分析
使用ANNOVAR软件[19]分析5个甜菜种质资源基因组中InDel位点的分布位置,以及不同长度的InDel位点在基因组上的分布情况。根据分析得出的结果确定可以开发InDel引物的合适位点。
2 结果与分析
2.1 甜菜品种的全基因组重测序分析
对5个甜菜种质资源进行全基因组重测序,测序后对测序质量进行分析,检查全基因组重测序结果是否符合规范,是否可以进行后续InDel位点检测与引物开发。分析结果表明测序错误率比较低(表2)。测序的错误率受到碱基质量、测量仪器、材料和原料等诸多因素的影响,产生误差的原因主要有以下几点:⑴测序仪器初期不稳定;⑵药剂的逐渐减少导致文库的测量质量降低;⑶DNA 被照射次数增多导致损伤增多。一般序列的前端和末端的错误率会偏高。

表2 测序数据的质量分析Table 2 Quality analysis of sequencing data
通过对5 个甜菜种质资源的全基因组重测序,共获得42.908 GB 的原始数据,经过过滤处理后得到42.849 GB 的有效数据。每个甜菜品种的原始数据在7 351.715~9 487.149 Mbp 的范围之内,Q20≥95.36%、Q30≥88.80%(表2)(高质量测序保证Q30>85%),G 和C 含量在36.79%~37.95%之间(表2)。结果表明,基因组测序数据量足够,C和G的分布在正常范围之内,测序质量良好,能够支持后续分析。
2.2 甜菜品种的基因组序列与参考基因组的比对分析
利用BWA软件(参数:mem-t 4-k 32-M)将5个甜菜品种的有效基因组数据与参考基因组比对,再利用SAMTOOLS 软件去除多余的重复序列。参考基因组的大小为566.550 Mbp(表3),由表4 可知,5 个甜菜品种的基因组平均测序深度最大为16.41%,最小为12.87%;单碱基覆盖度占比最大为95.79%,最小为94.62%;四碱基覆盖度占比最大为88.83%,最小为85.15%。比对后5 个甜菜品种与参考基因组一致的有效序列(去除重复序列)数据量为42.84 GB,有效序列的数量为2.71×109条,占参考基因组序列的99.84%。平均每个甜菜品种的有效数据量为8.57 GB,有效序列的数量为5.42×108条。结果表明,5个甜菜品种的基因组序列可用于后续的变异检测及相关分析。
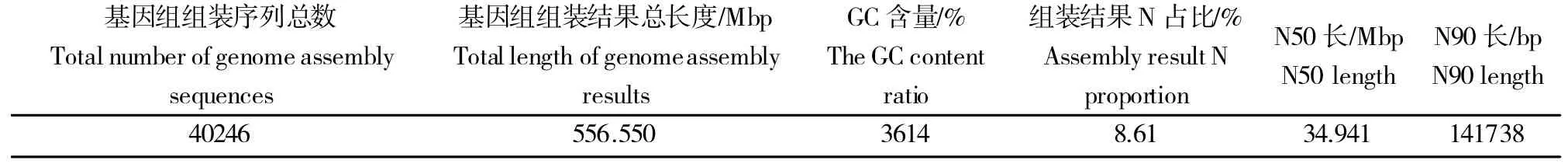
表3 参考基因组基本情况分析Table3 Analysisofreferencegenome

表4 测序深度及覆盖度分析Table 4 Analysis of sequencing depth and coverage
2.3 InDel序列长度及分布分析
利用SAMTOOLs(mpileup-m 2-F 0.002-d 1000)从5 个甜菜品种与参考基因组比对一致的序列中检测InDel序列(长度小于50 bp的插入与缺失片段),然后用ANNOVAR 软件对InDel序列的分布位置进行注释及分析(表5)。
通过InDel位点的注释分析,从5 个甜菜品种中分别发现了343 138、317 057、281 435、312 444和316 595个InDel位点(平均314 134.8),其数量和分布与参考基因组相似。在这5个甜菜品种中,InDel位点主要分布在基因间区,约占全部位点的55%,其次是基因区。在基因区内,大部分的InDel位点分布在内含子中,蛋白质编码区(Coding sequence,CDS)区中分布的InDel位点最少。

表5 InDel 位点的检测及注释Table 5 Detection and annotation of InDel locus
在这5 个甜菜品种全基因组中,InDel 位点的长度分布趋势也基本一致(图1)。InDel 位点的数量随着InDel 片段长度的增加而减少(基因区除外)。根据片段长度,可将InDel位点分为4 种类型,分别是长度为1 bp 的位点、长度为2~4 bp 的位点、长度为5~8 bp 的位点和长度超过9 bp 的位点。其中长度为1 bp 的InDel位点数量超过110 000 个(约占所有InDel的40%);长度为2~4 bp的InDel位点数量为20 000~80 000个;长度为5~8 bp的InDel位点数量约为10 000个;长度超过9 bp的InDel位点数量不足7 000个(图2)。

图1 基因编码区InDel长度分布Fig.1 InDel length distribution of gene coding region
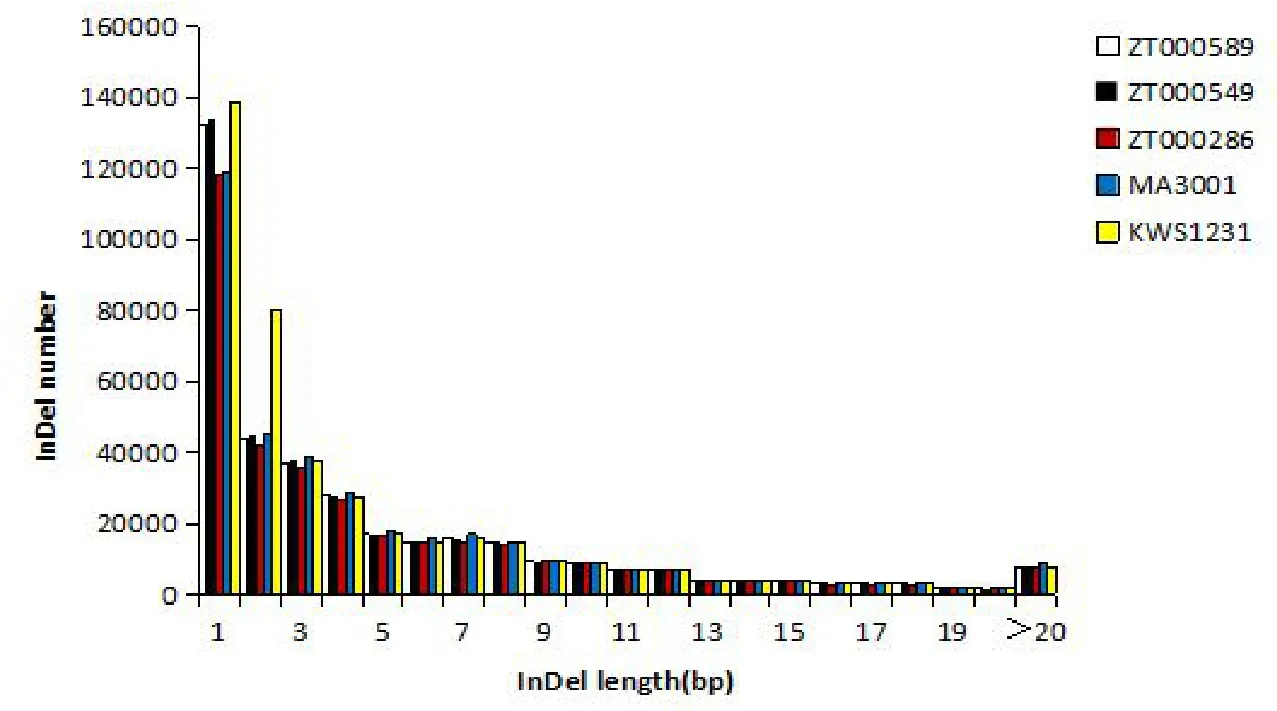
图2 全基因组InDel长度分布Fig.2 InDel length distribution in the whole genome
5个甜菜品种的InDel位点在CDS区的分布趋势和数量也与参考基因组相似,分别有4 828、4 810、4 473、4 975 和4 824 个InDel 位点,这些InDel 位点几乎都会引起编码蛋白质的显著变化。其中约有1 500 个InDel位点导致了插入或删除,这将插入或删除编码蛋白质的一个或几个氨基酸。大约有600~800 个InDel 位点导致了碱基移位,低于100个InDel位点导致了终止密码子的产生和缺失。导致移码突变或终止密码子的产生和缺失的InDel位点将大大改变编码蛋白质的序列。
用InDel位点的长度对不同长度InDel位点占位点总数的百分比作图,得到InDel位点在全基因组中的长度分布(图2),随着InDel 位点长度的增加,InDel 位点的数量将会减少。在基因编码区内,当InDel片段的长度为3 bp时,数量将远远超过其他长度的InDel位点(图1)。
3 讨论
3.1 甜菜品种基因组中InDel位点的分布
本研究使用Illumina测序平台对5个甜菜品种进行了全基因组重测序,并将全基因组序列分别与甜菜的参考基因组进行了比对,约有55%的InDel位点分布在基因间区,与其他物种的InDel位点分布基本一致。在基因区内,超过60%的InDel位点分布在内含子中,这些变异几乎不会影响基因表达和基因功能。如果InDel位点分布在CDS 区中,其编码蛋白质的氨基酸序列会发生改变,基因功能可能会发生改变甚至遭到破坏。如果InDel位点分布在启动子区域,基因的表达会增加或减少,这最终会改变植物的表型或适应性。分布在基因间区的InDel位点不会使基因表达发生如此大的变化,所以该区域中长度大于3 bp 的InDel位点最适合进行InDel 引物开发,其次是内含子区域中长度大于3 bp 的InDel位点,CDS 区域和启动子区域的InDel位点不适合进行引物开发。
3.2 甜菜品种基因组中InDel序列长度分布
InDel序列的长度各有不同,通常插入与缺失位点的数量会随着InDel长度的增加而减少(基因区除外),5 个甜菜品种的InDel 序列无论是在全基因组上的分布还是在CDS 区域的分布都符合这一情况。这一现象可能是因为植株基因间区较短,过长的插入或缺失会破坏与植物生存相关的基因,最终降低植物的存活率。当插入与缺失片段短时,对基因组的损伤较小,植株的存活率较高,因此这种变化保留的概率更高。在基因区内长度为3 bp 或其整数倍的InDel 位点的数量最多。最适宜开发InDel引物的是等于或大于3 bp 的InDel序列。将这5 个甜菜品种的基因组序列与参考基因组比对后发现,这5 个甜菜品种的基因组中分别有135 639、137 011、123 776、145 478 和135 040个InDel位点的长度超过3 bp,平均每MB数据中有238.78个长度超过3 bp的InDel位点。这些位点有适合用于开发高效的InDel引物。
4 结论
进行了5 个甜菜品种的全基因组重测序分析,Q20≥95.36%、Q30≥88.80%,基因组重测序数据量足够,C和G的分布在正常范围之内,测序质量良好,能够支持后续分析。
将5 个甜菜品种的基因组序列分别与参考基因组比对,经处理后得到42.84 GB 的无重复有效数据。通过InDel位点的注释分析可知,从5个甜菜品种中分别发现了343 138、317 057、281 435、312 444和316 595个InDel 位点。主要分布在基因间区,约占全部位点的55%,其次是基因区。在基因区内,大部分的InDel位点分布在内含子中,蛋白质编码区(Coding sequence,CDS)中分布的InDel位点最少。
InDel位点的数量随着InDel长度的增加而减少(基因间区除外),在基因区内大部分InDel位点的长度为3 bp或其整数倍,在基因间区内大部分InDel位点长度为1 bp。
5个甜菜品种的InDel位点在CDS区的分布趋势和数量也与参考基因组相似,分别有4 828、4 810、4 473、4 975和4 824个InDel位点,这些InDel位点几乎都会引起编码蛋白质的显著变化。
大于3 bp 的InDel位点适合进行InDel 引物开发。这些InDel位点的开发为进行遗传效应分析并鉴定甜菜种质资源提供了重要基础。
猜你喜欢
分子催化(2022年1期)2022-11-02
军事文摘(2022年16期)2022-08-24
今日农业(2021年11期)2021-08-13
昆明医科大学学报(2021年3期)2021-07-22
烟草科技(2021年6期)2021-06-24
小哥白尼(趣味科学)(2021年12期)2021-03-16
悦游 Condé Nast Traveler(2021年4期)2021-01-13
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
中国糖料(2020年1期)2020-01-15
