喹嗪并[3,4,5,6-ija]喹啉衍生物的合成及其光解水制氢性能
2020-07-20 02:06位春江徐良轩刘雪粉任晨超王天琦吴庆安罗书平
无机化学学报 2020年7期
位春江 徐良轩 刘雪粉 陈 浩 任晨超 王天琦 吴庆安 罗书平*,
(1浙江工业大学,绿色化学合成技术国家重点实验室培育基地,杭州 310014)
(2杭州师范大学钱江学院,杭州 310006)
能源和环境是当今人类面临的2个热点问题。太阳能作为一种清洁且可持续的能源,越来越受到人们的广泛关注[1]。目前人类能够直接利用太阳能的方法是通过一些半导体材料将光能转换成电能,但该方法的缺点是能量转化效率低。因此,为了更加充分地利用太阳能,将太阳能转换成化学能的方法逐渐受到关注。氢气具有能效高、无污染、零排放等优点,有望在未来取代传统的化石能源[2]。目前使用到的氢气大部分是从生物质、天然气或煤气化中产生,在生产过程中会对环境造成一定的污染,而电解水制氢的能耗比较高,因此通过太阳光催化分解水制氢是开发清洁和可持续氢能的理想途径之一[3-6]。
Fujishima和Honda于1972年在TiO2单晶电极上通过光催化实现了水分解产氢,证明了利用太阳能直接将水分解产生氢气这一方法的可行性,为光催化产氢的研究开辟了新的道路[7]。这一研究使光催化降解水制氢成为当今太阳能利用的研究前沿,在很短的时间内吸引了大量的学者,并取得了不错的研究成果。截止到目前,太阳光降解水产氢体系主要由光敏剂(PS)、水还原催化剂、电子牺牲剂3部分组成[8]。其中,光敏剂的主要作用是吸收光子然后将自身获得的能量传递给不能吸收光子的水还原催化剂。因此光敏剂是制约光解水产氢体系好坏的关键因素,研发高效、稳定、廉价、无毒的光敏剂是整个光解水体系研究的重要任务。最近几年,一些金属配合物由于具有较好的光物理性质,被广泛应用于光解水产氢体系中[8-10],如钌[11]、铑[12]、铱[13]、铂[14]、铜[15]等金属配合物。但这些光敏剂存在价格昂贵、储量稀少、稳定性差、有毒等缺点,这些缺点限制了它们在光解水产氢体系中的应用。
有机染料具有廉价易得、储量丰富、无毒或者毒性很小等特点[16-17]。有机染料最早是作为光敏化剂负载在一些半导体材料上,如TiO2、CdS以及ZnO等[18]。它可以有效拓展半导体吸收光谱响应范围,提高光解水制氢体系的产氢效率。因此有机光敏染料可以作为金属配合物的替代物应用于光解水产氢体系中。已经有文献报道将纯有机染料成功应用于光解水产氢体系,如氧杂蒽(荧光素及其衍生物)以及吖啶类[19-24]等。在这些报道中有机染料单独作为光敏剂,不需要负载在半导体材料上,吸收光谱响应范围广,产氢性能也比较好。此外,在2018年,我们课题组以三乙胺为电子牺牲剂,PdCl2+PPh3(1∶2,n/n)为制氢催化剂,以多咔唑取代苯腈的热活化延迟荧光(TADF)染料作为光敏剂,首次开拓了基于TADF光敏染料的高效光解水催化体系,其催化光解水产氢总转换数可达2 023[25]。因此,为了进一步研究光敏染料的制氢体系,我们设计合成了一系列多环喹啉类有机染料,并对其在可见光光催化降解水产氢体系中的光敏性能进行了研究。
1 实验部分
1.1 仪器与试剂
合成所用试剂均购自商业公司,所有溶剂都经过重蒸除水。紫外光谱分析使用瓦里安公司的CARY100紫外−可见−近红外分光光度计;NMR分析使用瑞士Bruker公司的Advance-Ⅲ核磁共振仪;荧光光谱分析使用SHIMADZU公司的RF-6000荧光分析仪;CV分析使用上海辰华的CHI660E电化学工作站;MS分析使用美国Thermo公司LCQ-advantage质谱仪;光催化反应所用光源为泊菲莱PLSSXE300UV 氙灯(辐照度:6 mW·m−2;波长:320~1 100 nm;输出功率:1.5 W)。
1.2 合 成
1.2.1 喹嗪并[3,4,5,6-ija]喹啉及其衍生物的合成
喹嗪并[3,4,5,6-ija]喹啉及其衍生物的合成路线如图1所示。

图1 喹嗪并[3,4,5,6-ija]喹啉衍生物的合成路线Fig.1 Synthesis routes of quinazino[3,4,5,6-ija]quinoline derivatives
1.2.2 化合物1和2的合成
原料化合物1(1-芳基吡啶三氟甲磺酸盐)和2(二芳基乙炔)以及所用催化剂的合成按照参考文献[26-27]进行。
1.2.3 不同取代基衍生物的合成
以3a为例,合成方法如下:氩气保护下,将试剂1(0.152 g,0.5 mmol)、2(0.196 g,1.1 mmol)、催化剂[Cp*RhCl2]2(0.015 g,0.025 mmol)和醋酸铜(0.399 g,2 mmol)加入史莱克管中,最后加入乙腈5 mL,将油浴锅温度设置为130℃,搅拌12 h。反应结束后将反应液冷却至室温,过滤,所得粗品经硅胶柱层析(V二氯甲烷∶V乙酸乙酯=5∶1)提纯。其余衍生物的合成均参照3a进行。
3a:浅黄色固体,收率70%。1H NMR(500 MHz,CDCl3):δ8.23(t,J=8.2 Hz,1H),8.05(d,J=8.2 Hz,2H),7.97(s,3H),7.41~7.34(m,16H),7.32~7.28(m,4H)。13C NMR(125 MHz,CDCl3):δ146.10,135.16,135.02,134.74,130.65,129.47,129.00,128.75,128.59,127.40,124.12。ESI-MS:[M−OTf]+m/z=508。
3b:黄色固体,收率69%。1H NMR(500 MHz,DMSO-d6):δ8.56(s,1H),8.06(d,J=8.3 Hz,2H),7.90(d,J=20.5 Hz,2H),7.63(s,1H),7.50(t,J=7.4 Hz,4H),7.47~7.42(m,4H),7.40(dd,J=10.2,3.4 Hz,4H),7.34(d,J=7.5 Hz,4H),7.31(d,J=8.7 Hz,3H)。13C NMR(125 MHz,DMSO-d6):δ162.91,160.95,145.20,141.91,141.80,135.13,133.92,132.64,131.41,130.29,129.14,127.65,126.68,124.97,124.16,116.01。ESI-MS:[M−OTf]+m/z=544。
3c:黄色固体,收率 71%。1H NMR(500 MHz,DMSO-d6):δ8.60(s,1H),8.53(s,2H),8.25(s,2H),7.92~7.91(m,4H),7.84(dd,J=4.5,3.4 Hz,4H),7.65(s,1H),7.51(s,4H),7.50(s,4H),7.41(s,2H)。13C NMR(125 MHz,DMSO-d6):δ152.87,145.03,143.22,141.19,139.88,138.03,133.41,132.80,130.26,129.13,127.47,126.68,125.56,124.40,124.21,118.33,111.58。ESIMS:[M−OTf]+m/z=558。
3d:棕黄色固体,收率75%。1H NMR(500 MHz,CDCl3):δ9.19(s,1H),8.61(s,1H),8.23(s,2H),7.68~7.68(m,1H),7.55(s,4H),7.52~7.52(m,4H),7.45(s,2H),7.33~7.32(m,8H),2.33~2.33(m,6H)。13C NMR(125MHz,CDCl3):δ159.20,146.30,142.45,140.72,136.06,132.36,132.32,131.81,129.43,129.24,128.76,127.47,127.42,126.24,125.22,124.93,124.13,21.29。ESI-MS:[M−OTf]+m/z=536。
3e:黄色固体,收率 70%。1H NMR(500 MHz,DMSO-d6):δ8.56(s,1H),8.52(s,2H),8.22(d,J=2.0 Hz,2H),7.86(s,1H),7.84(s,4H),7.51(s,4H),7.50(s,5H),7.01~7.01(m,4H),3.35(s,6H)。13C NMR(125 MHz,DMSO-d6):δ159.07,159.05,144.96,142.27,141.87,137.41,134.58,129.74,129.17,128.79,127.67,126.77,126.50,126.43,124.01,114.16,55.12。ESIMS:[M−OTf]+m/z=568。
3f:棕黄色固体,收率75%。1H NMR(500 MHz,CDCl3):δ8.23(t,J=8.2 Hz,1H),8.07(d,J=8.2 Hz,2H),8.01(d,J=6.7 Hz,2H),7.25(s,1H),7.25~7.13(m,8H),6.91(dd,J=8.4,3.8 Hz,8H),3.82(s,12H)。13C NMR(125 MHz,CDCl3):δ159.61,159.55,146.30,142.80,139.54,136.02,129.29,128.59,127.37,126.83,124.96,123.95,114.56,55.28。ESI-MS:[M−OTf]+m/z=628。
4a:棕色固体,收率 60%。1H NMR(500 MHz,DMSO-d6):δ10.53(s,1H),9.27(s,1H),8.55(s,1H),8.31(s,1H),8.24(s,1H),8.02(s,1H),7.22(dd,J=26.4,8.6 Hz,4H),6.99(dd,J=8.6,6.5 Hz,4H),3.37(s,6H)。13C NMR(125 MHz,DMSO-d6):δ159.01,158.97,146.35,143.95,141.08,140.77,134.87,132.49,130.64,128.89,127.29,126.90,126.46,123.99,121.95,118.41,114.12,55.12。ESI-MS:[M−OTf]+m/z=392。
1.3 光解水产氢实验
以3a为例,实验过程如下:光催化制氢体系先进行3次抽换气,排除体系里面含有的空气,使整个体系处于氩气保护。将3a(1.8 mg,3.5µmol)和二氯化钯(8.9 mg,5µmol)加入光反应瓶中,加入10 mL溶剂(VTHF∶VTEA∶VH2O=4∶3∶1;THF为四氢呋喃,TEA为三乙胺),用循环恒温槽将体系稳定在25℃,开启搅拌,反应体系由无色逐渐变为黄色。开启氙灯(不加滤光片)照射光反应瓶,并使用U型管收集体系产生的氢气,等到整个反应体系不再产生气体时,停止搅拌,关闭光源。同时每0.5 h观察记录一次产生的气体体积,并用气相色谱检测收集的气体。
1.4 光谱测试
取光敏剂3.5µmol,用四氢呋喃溶解后稀释至50 mL容量瓶中,在室温下进行光物理化学测试。
1.5 循环伏安测试
配制0.1 mol·L−1四丁基六氟磷酸铵的四氢呋喃电解质溶液,加入3.5µmol 3a,稀释到100 mL容量瓶中,使用三电极进行循环伏安电化学测试。循环伏安实验条件:工作电极为玻碳电极(d=2 mm),对电极为铂电极,参比电极为Ag/AgNO3电极(0.799 5 V vs NHE),室温,电解质是已经配好的 0.1 mol·L−1的四丁基六氟磷酸铵的四氢呋喃溶液,扫描速度100 mV·s−1,阶跃电位2.5 mV,调制时间0.05 s,间隔时间0.5 s,最高电位1.9 V(vs Ag/AgNO3),最低电位−2.0 V(vs Ag/AgNO3)。
2 结果与讨论
2.1 光解水制氢性能研究
为研究这类喹嗪并[3,4,5,6-ija]喹啉衍生物(简称喹嗪并喹啉)的光敏化性能,选择均相光解水产氢体系为测试平台,以3a为模板光敏剂,对体系的催化剂、溶剂等影响因素进行系统研究。
制氢催化剂是整个光催化制氢体系的重要组成部分,负责接收光敏剂的激发态能量然后催化分解水制氢,所以选择合适的催化剂对整个光解水产氢体系就显得尤为重要。以3a为光敏剂,分别对不同种类的催化剂进行了筛选。由表1可知,常用的非贵金属制氢催化剂如十二羰基三铁、联吡啶钴和镍等催化制氢性能较差;而贵金属钯具有较好的催化性能,其催化产氢的制氢总转换数(TON)可达142。其原因可能是喹嗪并喹啉光敏剂的激发态能量更加容易转移至钯催化剂,但我们没有观察到钯和光敏剂的配位作用。空白对照实验结果显示,当不使用任何催化剂时,光催化产氢体系几乎不会有气体产生。此外对催化剂二氯化钯的用量进行了优化,当催化剂用量为5µmol时TON值最高。由表1可知增加催化剂的用量并不能大幅度增强体系产氢性能。可能的原因是过量的二氯化钯催化剂干扰光敏剂发生还原淬灭过程,进而使整个体系的产氢性能下降,该现象和荧光淬灭实验结果一致。
此外,还研究了不同种类的溶剂对体系光催化制氢活性的影响,并对光敏剂3a在不同溶剂中的荧光强度进行了测试。结果如表2和图2所示,在光敏剂、催化剂、温度和光源都一致的情况下,仅改变体系的溶剂种类,其催化制氢效果发生明显变化。当溶剂为THF、TEA、水时,制氢效果最好,体系的产氢量为5.6 mL(TON=142);而当溶剂为乙醇、TEA、水时,体系的产氢量降为4.3 mL(TON=109),故改变溶剂对光催化降解水产氢体系的催化活性有明显的影响,这在已有的文献中有类似的报道[28-29]。根据相关文献,活性变化的原因是溶剂种类的改变使得溶剂的光物理性质发生了改变,故而对光敏剂和催化剂的光物理性质也有一定的影响[30];同时,溶剂也会影响光催化过程中产生的中间体稳定性,进而影响体系的电子转移速率以及效率[31],最终对体系的制氢活性产生影响。溶剂种类的荧光测试所得结论和产氢情况基本一致。

表1 光解水制氢中催化剂的性能Table 1 Performance of the catalysts in photolysis of water to produce hydrogen

表2 光解水制氢中溶剂的影响Table 2 Effect of the solvents in photolysis of water to produce hydrogen

图2 光敏剂3a在不同溶剂中的荧光发射图Fig.2 Fluorescence emission spectra of photosensitizer 3a in different solvents
以3a为光敏剂,研究其在单波长照射下制氢体系的产氢效率。分别用380、400、450 nm三种不同的波长进行照射,结果如图3所示,在380、400和450 nm波长下体系的产氢量最高分别为4.2、3.2和2.6 mL。

图3 光敏剂3a在不同波长的光照下的产氢量Fig.3 Hydrogen production of photosensitizer 3a under different wavelengths of light
根据优化的催化条件,着重研究了光敏剂的取代基效应。从制氢实验结果(表3)可以看出,不添加光敏剂的体系基本没有氢气产生,说明在这个光解水体系中,只有喹嗪并喹啉具有吸收光的光敏作用。当取代基为甲氧基时,光敏剂表现出很好的光敏活性,其中3e的光敏活性最好,制氢TON可达341。然而当取代基为氟或氰基时,喹嗪并喹啉光敏剂制氢性能大大下降,TON仅为51和61。可能的原因是吸电子基团的存在使得喹嗪并喹啉母核的电子云密度下降,不利于激发态电子转移。而给电子基团取代基则相反,根据相关文献报道,给电子基团的存在有利于电子从取代基转移到喹嗪并喹啉母核,提高母核的电子云密度,因此激发态的光敏剂可以很好地将电子转移给催化剂,实现催化质子还原产氢[32-33]。但是当甲氧基数量增加时,光敏剂的光敏活性有所下降,可能的原因是甲氧基数量的增加,使得3f的吸收光光谱发生红移,同时也使得光敏剂的荧光量子效率下降。

表3 光敏剂取代基对光解水制氢的影响Table 3 Substitute effect of the photosensitizer for photolysis of water to produce hydrogen
2.2 光谱表征
2.2.1 紫外和荧光表征
由图4和表4可知,四芳基取代喹嗪并喹啉3a~3f的可见光区的最大吸收峰主要位于420~460 nm之间,同时在400~425 nm附近有一个肩峰,可以归属于分子共轭体系的π→π*跃迁。二芳基取代吡啶并喹啉4a的最大吸收峰位于380 nm,在可见光区吸收较弱。表明随取代基增加和母核的增大,3a~3f具有更大的共轭π环,促使其吸收峰发生了明显红移。芳基上的取代基对化合物的吸光性能也有明显的影响,当取代基为吸电子的氟(3b)或氰基(3c)时,与没有取代基的3a相比,最大吸收峰几乎没有变动,但摩尔吸光系数明显下降。当取代基为强供电子甲氧基时,3a~3f的摩尔吸光系数几乎没有变动,但有明显红移。

图4 光敏剂3a~3f和4a的紫外吸收光谱图Fig.4 UV absorption spectra of photosensitizer 3a~3f and 4a
从图5和表4可知,所测光敏剂3a~3f的最大荧光发射波长位于450~550 nm之间。它们的荧光量子产率在0.27~0.48之间。吸电子取代基可提高喹嗪并喹啉化合物的光致发光效率,同时荧光发射峰不会位移;而供电子取代基可进一步提高荧光量子产率,同时荧光发射峰明显红移(最高可达26 nm),其中含2个甲氧基的3e荧光量子产率最高,Φ3e=0.48。4a的荧光强度最低,荧光量子产率也小,原因是4a的苯环数量较少,空间位阻效应较低,导致其不足以从结构上抑制荧光浓度淬灭不利因素的影响[35]。
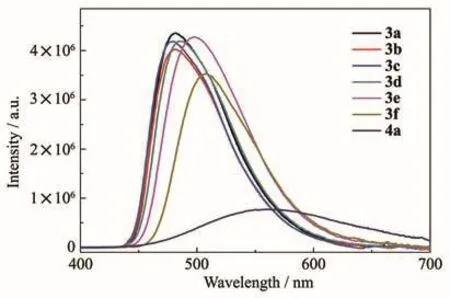
图5 3a~3f和4a的荧光光谱图Fig.5 Fluorescence emission spectra of 3a~3f and 4a
综合光谱表征分析和光解水产氢结果,光敏剂3e不仅在可见光区有很好的吸收峰,且在450~550 nm之间也有良好的荧光发射峰。光敏剂3e所表现出的良好的光敏活性主要归结于取代基的电子效应,甲氧基可以提高光敏剂在光催化体系中的光敏活性。但是从光敏剂的紫外吸收光谱图可以看出,随着甲氧基取代基数量增加,光敏剂的紫外吸收会发生明显红移,导致其光能利用效率降低,从而降低了制氢效果。

表4 光敏剂3a~3f和4a的光物理性质Table 4 Photophysical of photosensitizer 3a~3f and 4a
2.2.2 荧光淬灭
为了更好地研究光催化产氢体系中电子的转移过程,我们以光敏剂3e为例进行了荧光淬灭实验。将3e(3.5µmol)加入四氢呋喃中,然后分别使用不同浓度三乙胺以及催化剂二氯化钯作为淬灭剂,对3e的荧光进行研究。以I0/I对CQ作图,根据Sterm-Volmer方程(I0/I=1+KsvCQ)进行线性拟合,即可求出淬灭常数Ksv。方程中I0为未加任何淬灭剂时3e的荧光强度;I为加淬灭剂后3e的荧光强度;CQ为淬灭剂浓度。从图6和图7可以看出,当淬灭剂是三乙胺时,荧光淬灭效果非常明显,说明光敏剂3e在三乙胺存在下发生了还原淬灭,Ksv=23 L·mol−1(图 8);当淬灭剂是二氯化钯时,随着二氯化钯浓度的增加,光敏染料也发生了荧光淬灭,但淬灭效果不是十分明显。另外三乙胺用量远大于二氯化钯,故光敏剂3e在光解水产氢体系中主要的淬灭方式是还原淬灭。

图6 三乙胺对3e的荧光淬灭Fig.6 Fluorescence quenching of 3e by TEA

图7 二氯化钯对3e的荧光淬灭图Fig.7 Fluorescence quenching of 3e by PdCl2

图8 不同猝灭剂对光敏剂3e荧光淬灭的Sterm-Volmer图Fig.8 Sterm-Volmer plots of quenching PS 3e fluorescence with different quenchers
2.3 电化学表征
图9和图10分别为光敏剂3a~3f以及4a的循环伏安氧化电位以及还原电位图,光敏剂3a~3f的氧化还原电位相似,但取代基的影响仍然存在。氟取代基对还原电位影响较小,而氰基由于容易被还原,其还原电位明显下降。在氧化电位中,氟取代基能小幅度提高光敏剂的氧化电位,而其他取代基都是降低了氧化电位,以甲氧基取代基最为明显,与光敏剂的吸收峰检测结果相符。

图9 光敏剂3a~3g和4a的循环伏安图(相对于Ag/AgNO3的氧化电位)Fig.9 Cyclic voltammograms of 3a~3g and 4a(Oxidation potential vs Ag/AgNO3)

图10 3a~3g和4a的循环伏安图(相对于Ag/AgNO3的还原电位)Fig.10 Cyclic voltammograms of 3a~3g and 4a(Reduction potential vs Ag/AgNO3)
3 结 论
设计并合成了一系列喹嗪并[3,4,5,6-ija]喹啉衍生物,这些衍生物在均相光解水制氢中表现出良好的光敏活性,制氢TON最高可达341。经过吸收光谱以及荧光光谱和电化学研究,明确了喹嗪并[3,4,5,6-ija]喹啉衍生物的取代基效应,发现强供电子的甲氧取代基能明显提高喹嗪并[3,4,5,6-ija]喹啉衍生物的荧光量子效率(0.48),并合理解释了其光解水制氢活性好的原因。同时荧光淬灭实验表明,在均相光解水产氢体系中,光敏剂的主要淬灭途径是还原淬灭。
猜你喜欢
化工管理(2022年14期)2022-12-02
中国防痨杂志(2022年7期)2022-11-25
高等学校化学学报(2022年10期)2022-10-14
医学综述(2022年13期)2022-08-10
纺织检测与标准(2021年1期)2021-12-05
食品安全导刊(2021年20期)2021-08-30
煤气与热力(2021年6期)2021-07-28
中国食物与营养(2020年12期)2020-09-10
上海建材(2020年12期)2020-04-13
生物工程学报(2020年1期)2020-03-12