
腺嘌呤柱前衍生化-HPLC-FLD法测定3-氯-1,2-丙二醇的方法优化
2020-08-11 07:42胡金华李小敏胡志雄张维农齐玉堂张燕鹏
中国油脂 2020年8期
胡金华,杜 言,李小敏,胡志雄,2,张维农,2,齐玉堂,2,张燕鹏,2
(1.武汉轻工大学 食品科学与工程学院,武汉 430023; 2.大宗粮油精深加工教育部重点实验室(武汉轻工大学),武汉 430023)
氯丙醇类物质作为一种食品污染物近年来受到广泛关注,该类污染物在食品生产过程中中以3-氯-1,2-丙二醇(3-MCPD)生成量最多,因而3-MCPD被作为一种主要指标以反映食品加工过程中氯丙醇类物质的形成状况[1]。1995年,欧共体委员会食品科学分会对氯丙醇类物质的毒理作出评价,认为其是一种致癌物;FDA建议食物含3-MPCD水平不应超过1 mg/kg(干基);2001年,欧盟制定了限量标准,规定3-MPCD不得超过每日2 μg/kg的摄入量[2]。因此,开发高灵敏的3-MCPD分析方法对食品中3-MCPD污染残留进行监控具有重要意义。
目前,3-MCPD的检测方法主要有气相色谱法(GC)、气相色谱-质谱联用法(GC-MS)、毛细管电泳技术(capillary electrophoresis,CE)[3-4]、分子印迹(molecular imprinting,MIP)[5-6]等。其中基于GC的分析方法使用最为普遍, 为了便于检测,通常需要对3-MCPD 进行衍生化处理,常用的衍生化试剂主要有酮类(丙酮、丁酮、庚酮)[7]、硼酸类(丁基硼酸、苯硼酸)[8-11]、N,O-双三甲基-三氟乙酰胺(N,O-bis(trimethylsilyl)trifluoroacetamide,BSTFA)[12]、三氟乙酸酐(trifluoroacetic anhydride,TFAA)、七氟丁酰咪唑(heptafluorobutyrylimidazole,HFBI、heptafluorobutyric anhydride,HFBA)等[13-17]。该类方法在实际应用中仍存在不少问题, 如仪器设备条件要求高,分析所使用的氘代衍生化试剂比较昂贵, 以及衍生化试剂稳定性较差等。而采用CE和MIP等方法检测时,由于3-MCPD的光谱性能较差、相对分子质量小,使得检测灵敏度较低。
本课题组在综合分析3-MCPD结构特点的基础上,利用Malaprade反应,建立了一种腺嘌呤(Ade)柱前衍生化HPLC-FLD定量检测3-MCPD含量的分析方法,其检测原理如图1所示。
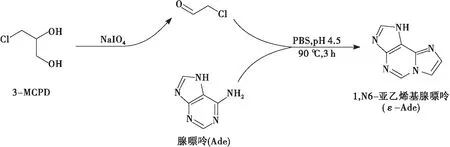
图1 腺嘌呤柱前衍生化-HPLC-FLD法检测3-MCPD原理[18]
3-MCPD结构中含有邻二醇基团,经高碘酸钠氧化裂解后可生成氯乙醛和甲醛,而氯乙醛与腺嘌呤(Ade)衍生化后生成强荧光物质——1,N6-亚乙烯基腺嘌呤(ε-Ade),可用HPLC-FLD检测,根据ε-Ade的检测结果可间接定性定量检测3-MCPD的含量。该法由于使用了荧光检测器,具有良好的检测灵敏度与选择性,另外因为其预处理、衍生化过程均在水相中进行,使得操作也极为简便。本文在前期工作基础上,对衍生化产物的荧光光谱特点进行了较深入的研究,对液相色谱流动相进行了优化,以期获得较佳色谱检测条件,进一步提高检测灵敏度与适应性。最后采用优化的检测条件对6种常见植物油样品中3-MCPD的含量进行了检测分析,以拓宽该方法的应用范围,同时为油脂生产企业与质量监督部门提供一种测定3-MCPD的新思路与新方法。
1 材料与方法
1.1 实验材料
1.1.1 原料与试剂
大豆油、玉米油、花生油、稻米油、菜籽油、山茶油,武汉某超市购买;3-MCPD(97%)、氯乙醛二乙缩醛(99.0%)、三水醋酸铅(99.9%),阿拉丁试剂公司;腺嘌呤(99.0%)、纯水(质谱级),百灵威试剂公司;浓盐酸(98%)、高碘酸钠、亚硫酸钠、氯化钠、磷酸氢二钠、柠檬酸、无水乙醇(色谱级)、甲醇、正庚烷、乙酸乙酯,国药集团化学试剂有限公司;pH 4.5的磷酸氢二钠-柠檬酸缓冲液(8.82 mL 0.2 mol/L Na2HPO4+ 11.18 mL 0.1 mol/L柠檬酸配制),腺嘌呤衍生化试剂用该缓冲液配制,现配现用。
1.1.2 仪器与设备
万分之一电子分析天平,梅特勒-托利多仪器有限公司;HHS电热恒温水浴锅,上海博迅实业有限公司;PHS-3C 雷磁pH计,上海精科电子有限公司;SK3300超声波清洗器,上海科导超声仪器有限公司;XW-80A微型旋涡混合仪,上海沪西分析仪器厂有限公司;Cary Eclipse型荧光分光光度计,美国瓦里安仪器有限公司;ESI/MS液相色谱-质谱联用仪,美国Thermo Fisher Scientific公司;安捷伦1260高效液相色谱仪,日本岛津公司; InertSustainTMC18 色谱柱(4.6 mm×250 mm×5 μm),日本GL Sciences公司。
1.2 实验方法
1.2.1 氯乙醛二乙缩醛水解制备氯乙醛[19]
量取5 mL(约32 mmol)氯乙醛二乙缩醛于50 mL圆底烧瓶中,加入15 mL 1.0 mol/L盐酸和5 mL无水乙醇,混匀后于70℃水浴中搅拌、回流反应2 h,冷却至室温后,用pH 4.5 磷酸氢二钠-柠檬酸缓冲液稀释、定容至32 mL,配制1.0 mol/L氯乙醛溶液,4℃冷藏保存备用。
1.2.2 3-MCPD溶液的高碘酸钠氧化裂解预处理与荧光衍生化
移取3.0 mL 3-MCPD溶液于5 mL离心管中,加入0.15 mL 250 mmol/L高碘酸钠溶液,避光反应30 min后,加入0.15 mL 250 mmol/L亚硫酸钠溶液反应20 min,取2.2 mL处理过的溶液,加入0.2 mL 16.0 mg/mL腺嘌呤溶液,在90℃下反应3 h,然后冷却至室温,经0.22 μm有机相滤膜过滤后进HPLC-FLD检测。
1.2.3 HPLC-FLD分析条件
InertSustainTMC18 色谱柱(4.6 mm×250 mm×5 μm),流动相为甲醇/碳酸钠-碳酸氢钠缓冲液 (pH 9.0) (体积比20∶80)溶液,流速0.5 mL/min,进样量20 μL, 紫外检测波长280 nm,最大激发波长236.0 nm,最大发射波长 405.0 nm。
1.2.4 衍生化产物的荧光光谱及影响因素研究
取2.0 mL稀释至0.1 mol/L的氯乙醛标准溶液,加入0.5 mL 0.1 mol/L腺嘌呤溶液,按1.2.2方法衍生化处理后,使用HPLC于紫外波长280 nm处进行分离(甲醇-水体系流动相,体积比15∶85),在衍生化产物的色谱峰处(17.5 min)将其流出液接出,接出液稀释后采用荧光分光光度计测定荧光激发光谱和发射光谱。为了研究HPLC分析条件下流动相pH、有机溶剂体积分数、离子强度对衍生化产物荧光性能的影响,将接出液取0.1 mL,分别加入不同溶液2.0 mL,混合均匀,以调节接出液的pH、有机溶剂甲醇体积分数和离子强度,然后扫描其激发光谱与发射光谱,记录最佳测定波长条件下的荧光强度,并绘制相应变化规律曲线。
1.2.5 方法评价
1.2.5.1 可行性评价——检测体系背景与可能干扰物的影响
背景对照实验处理方法:磷酸盐缓冲液体系按1.2.2方法以纯水代替3-MCPD溶液、磷酸盐缓冲液代替腺嘌呤溶液进行处理;腺嘌呤标准溶液(不加热)和腺嘌呤标准溶液(加热)体系则分别按1.2.2 方法以纯水代替3-MCPD溶液、腺嘌呤标准溶液加入后不加热或90℃加热反应3 h。
干扰物比较实验处理方法:1.0 μg/mL甲醛、1.0 μg/mL 乙醛、200.0 ng/mL氯乙醛标准溶液分别量取2.2 mL,加入0.2 mL腺嘌呤溶液衍生化反应;纯水、1.0 μg/mL甘油、1.0 μg/mL 1,2-丙二醇溶液与80.0 ng/mL 3-MCPD标准溶液则按1.2.2方法的步骤进行处理。
1.2.5.2 方法的定量性能评价
配制1.0~500.0 ng/mL 的3-MCPD标准溶液,按1.2.2方法进行衍生化处理后,采用1.2.3的分析条件进行检测,以ε-Ade的峰面积为纵坐标,3-MCPD质量浓度为横坐标绘制标准曲线,进行线性回归得标准曲线回归方程。
1.2.6 食用油中游离3-MCPD的测定
1.2.6.1 食用油样品加标实验
取150 mL油样溶于300 mL正庚烷,加入100 mL氯化钠溶液(0.01 g/mL),慢速搅拌萃取0.5 h后,静置分层并弃去下层水相,重复处理4次后,各取上层油相30 mL,分别加入2.5、5.0、10.0、20.0、40.0、80.0 mg/L 3-MCPD标准溶液(乙酸乙酯作溶剂)10 μL,涡旋混匀作为加标样品,加入6.0 mL 0.01 g/mL氯化钠溶液后,磁力搅拌20 min,静置分层后分出下层水相3.0 mL待测。
1.2.6.2 食用油样品中3-MCPD的萃取处理
准确量取32.0 g食用油(体积约为35 mL)置于150 mL 三角瓶中,加入70.0 mL正庚烷进行稀释。搅拌均匀后,取30.0 mL混合油样置于100 mL烧杯中,加入6.0 mL 0.01 g/mL氯化钠溶液后,磁力搅拌20 min,静置分层后分出下层水相3.0 mL待测,每种食用油准备3组平行处理样品。
1.2.6.3 食用油中游离3-MCPD氧化裂解预处理与荧光衍生化
移取3.0 mL油脂萃取的下层水相溶液于5 mL离心管中,加入0.15 mL 250 mmol/L高碘酸钠溶液,避光反应30 min后,加入0.15 mL 250 mmol/L亚硫酸钠溶液反应20 min,再加入275 mmol/L的醋酸铅溶液0.15 mL,涡旋振荡混匀,5℃静置10 min后,3 000 r/min离心10 min,取2.2 mL上层清液,按1.2.2方法进行荧光衍生化,再按1.2.3条件进行HPLC-FLD测定分析。
1.2.7 统计分析
所有数据均采用Origin 6.0绘图软件处理。
2 结果与分析
2.1 衍生化产物ε-Ade的荧光光谱及影响因素优化
2.1.1ε-Ade的荧光光谱
荧光检测波长的设定对于HPLC-FLD方法的检测灵敏度至关重要。为此,本实验按1.2.4方法,将较高浓度条件下制备的衍生化溶液在280 nm下分离后,接出ε-Ade色谱峰的流出液,稀释后进行荧光光谱测定,结果见图2。从图2可以看出,ε-Ade的最佳荧光发射波长为405 nm,而激发光谱则出现236 nm和276 nm两处激发峰,并且276 nm处的荧光强度大于236 nm处的荧光强度,这可能与ε-Ade 在溶液中的存在形态有关,为了对这一现象深入探索,本实验研究了pH与ε-Ade的荧光强度之间的变化规律。

图2 ε-Ade的荧光激发光谱(实线)和发射光谱(虚线)
2.1.2 pH对ε-Ade荧光性能的影响
按1.2.4方法,用pH 2.2~13.0的缓冲液(磷酸氢二钠-柠檬酸缓冲液(pH 2.2~8.0)、碳酸钠-碳酸氢钠缓冲液(pH 9.0~11.0)、磷酸氢二钠-氢氧化钠缓冲液(pH 12.0)、氯化钾-氢氧化钠缓冲液(pH 13.0))调节色谱峰流出液pH,测定在最大发射波长405.0 nm处的荧光激发光谱,结果见图3。
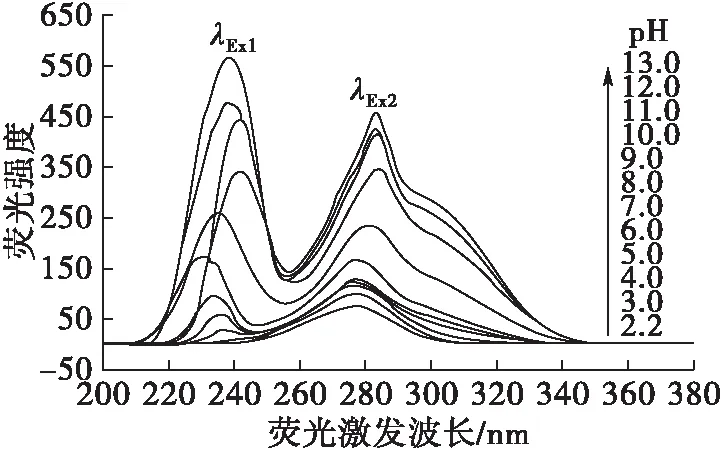
由图3可见:溶液pH较低(pH<4.0)时,基本上只出现一个较弱的激发峰(λEx= 276.0 nm),这与文献[20]报道的激发波长基本一致,随着pH的递增,在236.0 nm波长处逐渐出现另一个激发峰,形成双峰形态,并且两峰的荧光强度均随着pH的升高而增强,236.0 nm处的峰强增强速度大于276.0 nm处的激发峰,pH 为8.0时,两峰强基本相等,此后,随着pH的继续升高,236.0 nm处峰强均大于276.0 nm处峰强。产生该现象的原因可能与ε-Ade在溶液中的存在形态有关,当pH较低(pH<4.0)时,ε-Ade分子中6位N原子中的n电子质子化,质子化的ε-Ade只在276.0 nm附近产生一个激发峰,其共轭程度减弱导致相应激发波长均出现一定程度的蓝移;随着pH的升高,ε-Ade逐渐转变为中性形态,共轭程度不断增强,荧光强度随之逐渐增强,另外,中性ε-Ade呈现两个激发峰,其中236.0 nm处激发峰变化较为剧烈,可能为中性ε-Ade特征激发峰。
在HPLC分析过程中,碱性样品在硅胶基质色谱柱上进行分离时易发生拖尾现象,增强流动相的pH可以抑制硅羟基的电离,从而减少该现象的发生;另外,流动相的pH对易电离物质在HPLC分析中的保留也会产生重要影响。为了提高本方法检测灵敏度,根据以上实验结果的分析,本实验选择具有一定耐碱性的色谱柱InertSustainTMC18进行测定,其pH耐受范围为1.0~10.0,保守起见以下实验选择pH 9.0对分析条件进行优化,该pH下激发波长236.0 nm处的荧光强度稍大于276.0 nm处的,因此优选激发波长236.0 nm,发射波长则选择405.0 nm。
2.1.3 流动相甲醇体积分数对ε-Ade荧光性能的影响
HPLC分离过程中,流动相中有机相的比例不仅会改变其洗脱强度,还会对溶质的解离状态产生影响,为此,本实验对流动相中甲醇的体积分数与ε-Ade 荧光强度之间的关系进行了探索。按1.2.4方法控制流动相pH为9.0,调节色谱峰流出液甲醇体积分数为0~40.0%,在激发波长236.0 nm条件下测定相应荧光强度,结果如图4所示。

图4 流动相甲醇体积分数对ε-Ade荧光性能的影响
由图4可见,随着甲醇体积分数的升高,ε-Ade的荧光强度不断增强,甲醇体积分数为40.0%时的荧光强度较5.0%时增强约1倍。可能是因为流动相中甲醇体积分数升高时,ε-Ade的解离抑制作用不断增强,另外,甲醇对于中性ε-Ade具有一定增溶性,从而使得体系中ε-Ade中性形态比例增加所致。考虑到保留时间过短会影响方法的准确度与抗干扰能力,在权衡检测灵敏度与保留时间的基础上,最终确定流动相中甲醇体积分数为20.0%,该条件下ε-Ade保留时间为12.1 min。
2.1.4 离子强度对ε-Ade荧光性能的影响
在HPLC分离过程中,流动相中无机盐的离子强度一定程度上也会影响样品的解离状态、质子化程度,从而对其色谱峰的对称性、保留性能及荧光强度、检测灵敏度产生一定程度的影响。为此,本实验控制流动相pH为9.0(1.0 mL 0.1 mol/L Na2CO3+9.0 mL 0.1 mol/L NaHCO3)、甲醇体积分数20%,按1.2.4方法添加适量NaCl使其在色谱峰流出液中的浓度分别为0.00~1.00 mol/L,在激发波长236.0 nm条件下测定相应荧光强度,结果如图5所示。
由图5可见,增强流动相的离子强度,对于ε-Ade具有一定的荧光猝灭效应,随着体系中离子强度的增加,荧光强度不断降低,NaCl浓度为1.00 mol/L时的荧光强度相比未添加NaCl时降低约10.33%。通常,对于易解离的样品来说,增强溶液的离子强度将会抑制其解离,使质子化程度降低,对于分子态发光样品而言,荧光强度将有所增强。本实验条件下的结果似乎与该规律有所相悖。可能是因为在pH 9.0、甲醇体积分数20%的条件下,ε-Ade已基本处于分子态,荧光性能处于较优化状态,继续增强离子强度时,盐析效应所带来的荧光增强作用较小,而分子间距离缩短带来的碰撞猝灭效应反而更明显;另外,氯离子的重原子效应也可能会产生一定的荧光猝灭作用。因此,为了提高检测灵敏度,本实验条件下不宜在流动相中添加额外的无机盐来增强离子强度。
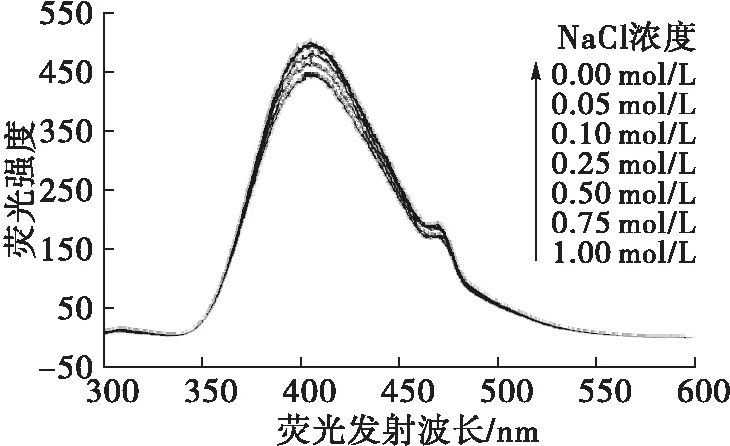
图5 NaCl浓度对ε-Ade荧光性能的影响
2.2 检测方法的评价
2.2.1 方法选择性与抗干扰性
为了评价方法检测的选择性以及样品基质中可能存在的干扰物对该方法准确度的影响,实验选择了甲醛、乙醛以及氧化裂解后可生成的相关物质甘油、乙二醇作对照,并对体系本身的一些成分按1.2.5.1方法进行处理并检测,得到HPLC谱图,结果如图6所示。

注:a.磷酸盐缓冲液;b.腺嘌呤标准溶液(不加热);c.腺嘌呤标准溶液(加热);d.纯水;e.1.0 μg/mL 甘油;f.1.0 μg/mL 1,2-丙二醇;g.1.0 μg/mL 甲醛;h.1.0 μg/mL 乙醛;i.80.0 ng/mL 3-MCPD;j.200.0 ng/mL 氯乙醛。
由图6可见:衍生化试剂Ade与衍生化产物ε-Ade 的色谱峰能够很好地分离,其保留时间分别为8.50 min和12.10 min;体系本身的成分如磷酸盐缓冲液、腺嘌呤标准溶液(加热或不加热)无ε-Ade的色谱峰出现,常见裂解产物甲醛、乙醛虽然质量浓度远大于3-MCPD,其色谱图中亦无ε-Ade的色谱峰出现;纯水、甘油、1,2-丙二醇经氧化裂解处理后进行衍生化,其色谱图中在目标峰处呈现极其微弱的小峰,为了探究该峰产生的原因,实验以质谱级纯水为样品,改变1.2.2方法中高碘酸钠氧化裂解后的处理程序,发现不添加还原剂亚硫酸钠时该峰比添加之后大很多,而添加亚硫酸钠处理之后,继续添加醋酸铅处理(添加0.15 mL 275 mmol/L 的醋酸铅溶液),将体系中高碘酸、碘酸根沉淀除去可以将该峰抑制至最低。这一现象说明,该处空白峰的出现可能是因为残存的高碘酸具有较强的氧化性,可以将衍生化试剂Ade氧化产生疏水性与ε-Ade相近的物质所致。甘油、1,2-丙二醇在目标峰处与纯水峰高一致,说明该干扰情况主要来源与水样一致。综上所述,衍生化试剂与3-MCPD氧化裂解产物氯乙醛的反应选择性非常高,基于该反应的检测方法抗干扰性好、灵敏度高。
2.2.2 方法定量性能
按1.2.5.2方法得到纯水体系下3-MCPD定量标准曲线方程为y=5.046x-12.842(y为峰面积;x为3-MCPD的质量浓度,ng/mL),线性回归系数(R2)为 0.997,说明线性关系优良。按信噪比3和10可得方法的检测限与定量限分别为0.06、0.20 ng/mL。与文献[12]报道的GC-MS/MS方法检测限相近,但远低于Matthew等[21]的GC-ECD法测定水样中3-MCPD的结果。
取20.0、100.0 ng/mL 3-MCPD样品2 mL,分别加入50、100、200 μL 800.0 ng/mL 的3-MCPD标准溶液,按1.2.2方法处理后检测,得到ε-Ade的实际峰面积,然后根据标准曲线计算的理论值统计加标回收率。结果表明,所有样品的加标回收率均在93%以上,平均加标回收率为96.12%,相对标准偏差在2.51%~3.48%之间,说明该方法的准确度与精密度很高。
分别取5.0、20.0、50.0 ng/mL 3-MCPD样品,按1.2.2方法处理后检测,以日内5次测定和连续5 d测定的方式对方法稳定性、重现性进行评价。结果表明,日内测定相对标准偏差为2.8%~4.6%,日间测定相对标准偏差为3.2%~5.6%,说明该方法的稳定性好,重复性高。
2.3 食用油中游离3-MCPD的测定
2.3.1 食用油中3-MCPD氧化裂解预处理条件优化
由于3-MCPD与油脂中各组分的极性相差较大,极易溶于水,所以采用简单的液-液萃取方法即可实现对其分离预处理。为了避免萃取中产生乳化现象,可采用NaCl溶液(0.01 g/mL)作为水相进行萃取。但是实验发现增加水相盐浓度后,出现空白溶液的目标干扰峰增大的情况。为此,按1.2.2方法对0.01 g/mL NaCl溶液的氧化裂解预处理条件进行比较优化,处理方法参数设置见表1,不同处理方法所测得的对应空白溶液HPLC谱图如图7所示。
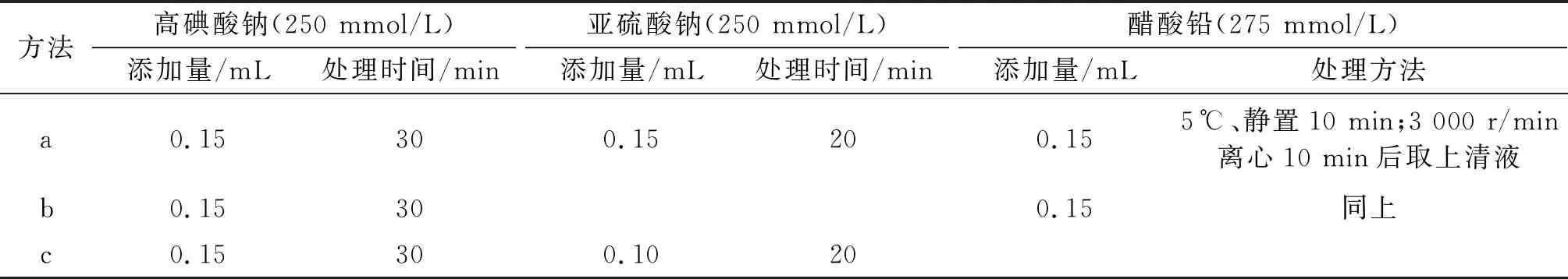
表1 降低空白峰的3种方法
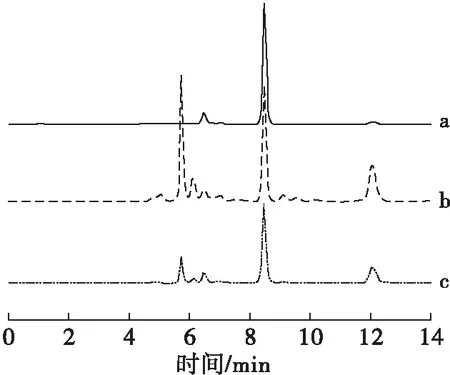
图7 3种降低空白峰的方法结果比较
从图7可以看出,按c方法进行处理时,在目标峰处会出现相对较高的背景干扰,这可能是由于离子强度增大时,亚硫酸钠对于高碘酸钠的还原反应会受到一定程度的抑制,残留的痕量高碘酸根离子含量增强,使得ε-Ade的干扰产物有所升高所致,如果在亚硫酸钠处理后,加入醋酸铅使体系中的氧化性阴离子沉淀脱除后,干扰峰减弱至可忽略不计的水平,所以对于油脂样品中3-MCPD的检测,采取了方法a对液-液萃取后的水相体系进行处理,具体操作步骤如1.2.6.3方法中所述。
2.3.2 食用油中3-MCPD定量测定方法评价
采用0.01 g/mL NaCl溶液代替纯水作为3-MPCD萃取溶剂后,其定量标准曲线也会因此有所差异,为此,需要用0.01 g/mL NaCl溶液配制1.0~500.0 ng/mL 的3-MCPD标准溶液,按1.2.6.3方法进行预处理后进行HPLC-FLD测定,再根据1.2.5.2 方法绘制相应标准曲线,所得定量标准曲线方程为y=4.33x+2.99 (R2=0.999 9)。按1.2.6.1 方法进行加标回收率测定,可得加标回收率为90.11%~102.70%,说明该方法标准曲线线性关系良好,准确度与精密度高,可用于食用油中游离3-MCPD的检测。
2.3.3 实际食用油样品检测
本实验以6种常见植物油样品为例进行了相关测定,按1.2.6.2方法对其液-液萃取,萃取所得下层水相按1.2.6.3方法进行预处理、HPLC-FLD检测,6种常见植物油HPLC谱图如图8所示。
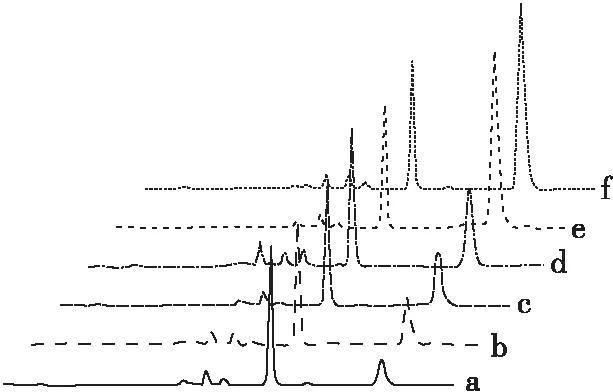
注:a.玉米油;b.山茶油;c.稻米油;d.大豆油;e.花生油;f.菜籽油。
由图8可见,采用该方法对6种常见植物油中游离3-MCPD检测时,谱图中衍生化产物峰形对称,基质干扰少,目标物检测选择性好。按上述定量标准曲线进行定量分析,玉米油、山茶油、稻米油、大豆油、花生油、菜籽油中3-MCPD的含量分别为6.28、10.53、12.17、17.70、59.99、89.75 μg/kg,3次重复测定的相对标准偏差为0.32%~4.63%。不同油脂3-MCPD含量之间差异较大,这可能与各种油料的理化特性以及精炼工艺处理方式不同有关。
3 结 论
本文在前期研究的基础上,对3-MCPD经高碘酸氧化裂解、腺嘌呤衍生化后产物的荧光光谱特点进行了深入探讨,并对影响衍生化产物荧光性能的因素进行了研究,对HPLC-FLD检测条件进行了优化。另外,在优化的检测条件下,对植物油样品中3-MCPD的检测进行了应用评价。结果表明:在流动相pH 9.0、流动相甲醇体积分数20%、流动相中不添加额外的盐离子、激发波长236.0 nm、发射波长405.0 nm时,对于水相体系中3-MCPD具有更高灵敏度(检测限与定量限分别为0.06、0.20 ng/mL);用于植物油中游离3-MCPD测定时,以 0.01 g/mL的NaCl溶液作为萃取水相,在3-MCPD 氧化裂解后的高碘酸钠处理过程中,加入1.1倍的醋酸铅溶液可以抑制空白峰的干扰。将其应用于6种常见食用植物油样品的测定,结果表明该方法准确度高、重现性好(RSD在0.32%~4.63%之间),样品谱图显示基质干扰很小,选择性高。本方法可为油脂生产企业与质量监督部门提供一种3-MCPD测定的新思路与新方法。
猜你喜欢
农业工程学报(2022年5期)2022-06-22
世界科学技术-中医药现代化(2021年5期)2021-11-05
昆明医科大学学报(2021年8期)2021-08-13
天津医科大学学报(2021年3期)2021-07-21
中国科技纵横(2021年24期)2021-03-02
蚕桑通报(2020年1期)2020-07-10
湖北科技学院学报(医学版)(2020年1期)2020-01-08
中央民族大学学报(自然科学版)(2018年3期)2018-11-09
分析化学(2018年12期)2018-01-22
分析化学(2014年8期)2014-09-02
