
Nanopore 单分子测序基因组结构变异分析流程比较
2020-08-13 12:48王曦路
世界最新医学信息文摘 2020年54期
王曦路
(复旦大学 生命科学学院,上海 200000)
0 引言
结构变异(Structural variant,SV)包括插入,缺失,重复,倒位和易位(一般定义为大于50bp)[1-3]。在基因组中存在的遗传变异形式中,结构变异仍然是其功能影响最难以解释的变异之一。一直以来基因组结构变异被认为与表型多样性、人类疾病、基因多样性以及大规模染色体进化等有关,但他们的影响仍未完全清楚。目前对于结构变异的功能影响的研究主要来源于人类疾病研究方面[1-5]。
从Sanger 测序到下一代测序(NGS),可以从测序数据中收集的信息量和丰富程度大大增加,同时测序成本急剧下降[6]。测序技术的进步使得对SNP 以及小的插入和缺失等变异的检测和分析取得了长足的进展,但受限于读长,使用NGS 进行SV 检测仍然面临许多困难[7]。以Oxford 开发的Nanopore 测序技术[8]以及Pacific Biosciences(PacBio)开发的SMRT 测序技术为代表的单分子测序技术的出现使得长读长测序成为可能。借助单分子测序技术,近期已有多种遗传病找到了相关SV。如:双相情感障碍和精神分裂症[9],家族性皮质肌阵挛性震颤伴癫痫(familial cortical myoclonic tremor with epilepsy,FCMTE)[10],神经元核内包涵体病(Neuronal intranuclear inclusion disease, NIID)[11-13]等。但单分子测序技术仍然存在一定的局限性,最主要的是较高的测序错误率和更加昂贵的成本[14]。因此,使用适当的分析方法以及覆盖度进行检测在减少错误和控制成本上就显得尤为重要。
目前针对Naopore 测序数据可以使用的比对软件主要有以下几种:NGMLR[15],BWA-MEM[16],Graphmap[17],Minimap2[18]。SV 发 现 软 件 有NanoSV[19]和Sniffles[15]。Genome in a Bottle(GIAB)联盟发布了针对NA12878 基因组的高可信度SV 集合(2676 个缺失SV 以及68 个插入SV)。这是一个由不同平台进行深度测序得到的集合,并在家系中验证准确率为99.7%,可以对数据分析流程进行性能验证[20]。我们评估了四个比对软件和两个SV 发现软件的组合性能。这将对Nanopore 测序在临床及科研上的SV 检测提供一定的依据。
1 材料与方法
1.1 研究使用的Nanopore 数据集。本研究使用的数据集来自GIAB 联盟发布的基于Nanopore 测序平台的NA12878基因组测序数据(https://github.com/nanopore-wgsconsortium/NA12878)[21]。该数据由多家实验室分别测序获得。在获得Fastq 数据后,以人参考基因组(NCBI build 37)作为参考序列(与高可信度SV 集合保持一致),分别以不同的覆盖倍数(2-30×)进行随机抽样,将抽样得到的Fastq 作为起始数据进行后续研究。
1.2 数据比对和发现SV。分别使用NGMLR(默认参数)[15],BWA-MEM(bwa mem –x ont2d -M)[16],Graphmap(默认参数)[17]和Minimap2(默认参数)[18]将抽样的fastq 数据比对到人参考基因组(NCBI build 37)上,产生SAM 文件。
之后分别使用NanoSV[19]和Sniffles[15]进行SV 检测,Sniffles 需要修改参数(最小reads 支持修改为2)以增加SV 检测的灵敏度,如图1 所示。
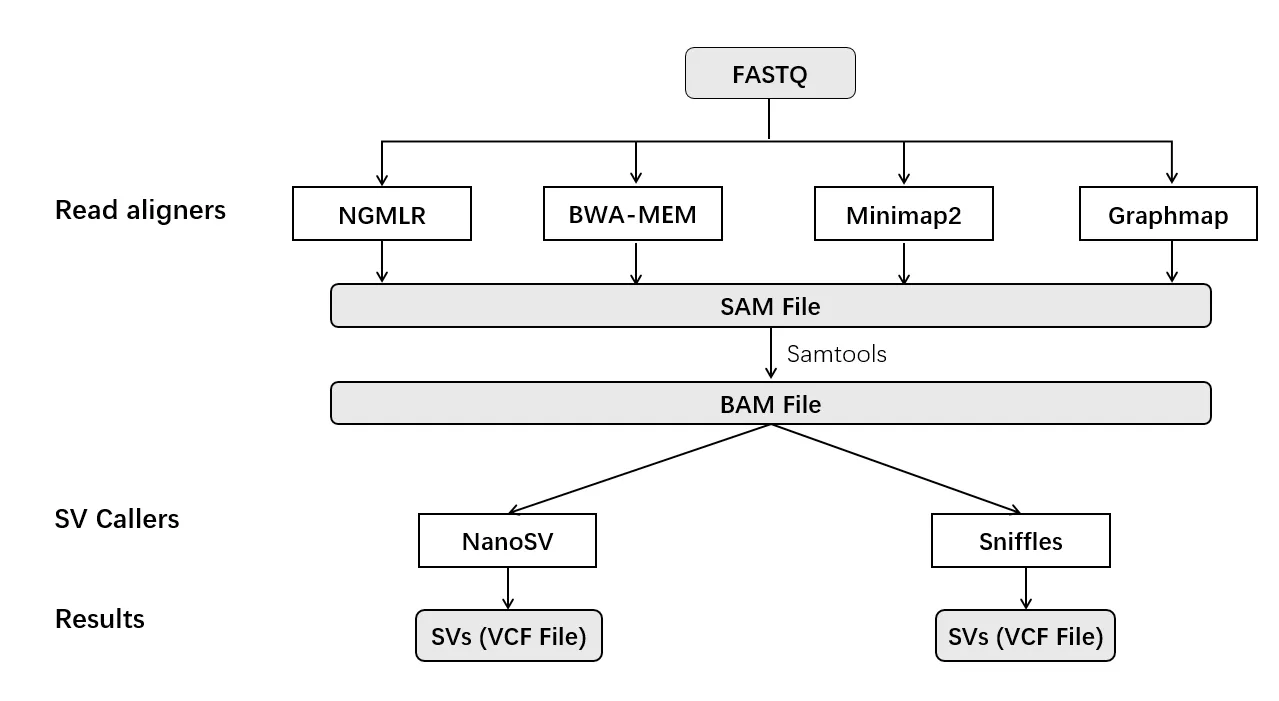
图1 数据比对和发现SV 流程
1.3 性能评估。分别获取各分析流程的各覆盖度下的SV 集合与高可信度SV 集合的共识SV。以评判其准确度(检测到的标准SV 中的SV 在该流程的得到的所有SV 中的百分比)和召回率(该流程检测到的SV 在标准SV 中的百分比)。比较两个SV 是否相同时,缺失SV 在基因组上显示为一个区域,而插入SV 仅有一个断点坐标,因此需要使用不同的标准。对于缺失SV,两个缺失之间的重叠区域超过50%则认为它们是相同的。插入SV 的判断在之前的研究中标准差异较大,如果两个插入SV 之间相距不超过500bp,则认为两个插入相同[22]。
2 结果
2.1 各流程在各覆盖度下的SV 发现数量。为了确定Nanopore 数据中SV 检测的最佳覆盖度,我们使用抽样的2×,4×,6×,8×,10×,12×,15×,20×,25× 和30×,在每个覆盖度下分别使用NGMLR,BWA-MEM,Graphmap 和Minimap2 进行比对,之后分别使用NanoSV和Sniffles 进行SV 发现。各分析流程各覆盖度下发现的SV数量如图2 所示。随着覆盖度的增加,SV 的数量都在持续增加,这可能是由于Nanopore 本身的测序错误率较高导致的。但是除Minimap2 分析流程外,在超过20×之后,SV 的增量均有明显的的降低,尤其是使用NanoSV 发现SV 的流程中更为显著。
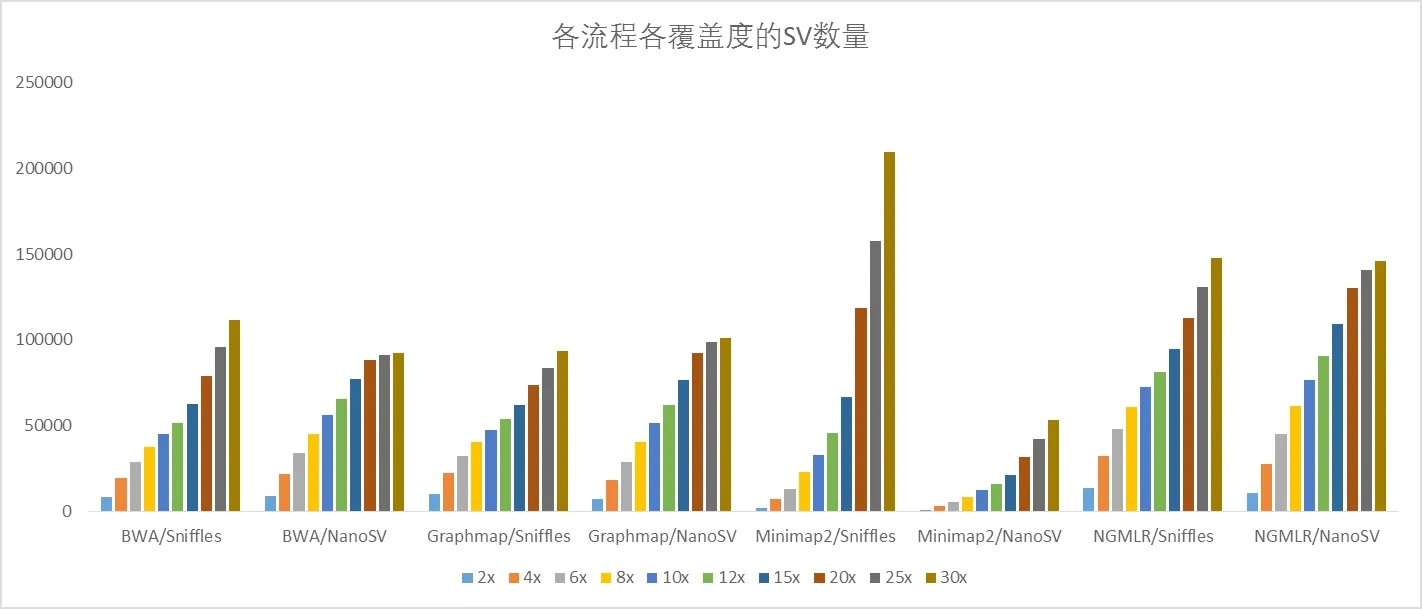
图2 各分析流程各覆盖度下发现的SV 数量
2.2 不同流程之间的性能差异。在所有流程中的召回率都是随着覆盖度的升高而增加,20×之后趋势变缓。
30× 覆盖度下对于缺失SV 的召回率最高的是NGMLR/NanoSV 流 程( 召 回 率:96.936%, 准 确 率:2.368%);而召回率最低的是Minimap2/Sniffles(召回率:18.984%,准确率:0.247%)。20×覆盖度下,召回率最高为NGMLR/NanoSV(召回率:94.918%,准确率:2.463%),与30×下差异不大。
30× 覆盖度下对于插入SV 的召回率最高的是Graphmap/NanoSV 流程(召回率:80.882%,准确率:0.119%);而召回率最低的是Minimap2/Sniffles(召回率:19.118 %,准确率:0.006%)。20×覆盖度下,召回率最高为Graphmap/NanoSV(召回率:77.941 %,准确率:0.122%),与30×下差异不大。
由此,结合成本和召回率考虑,选择以20×左右的覆盖度作为标准较为合适详情加图3,图4。
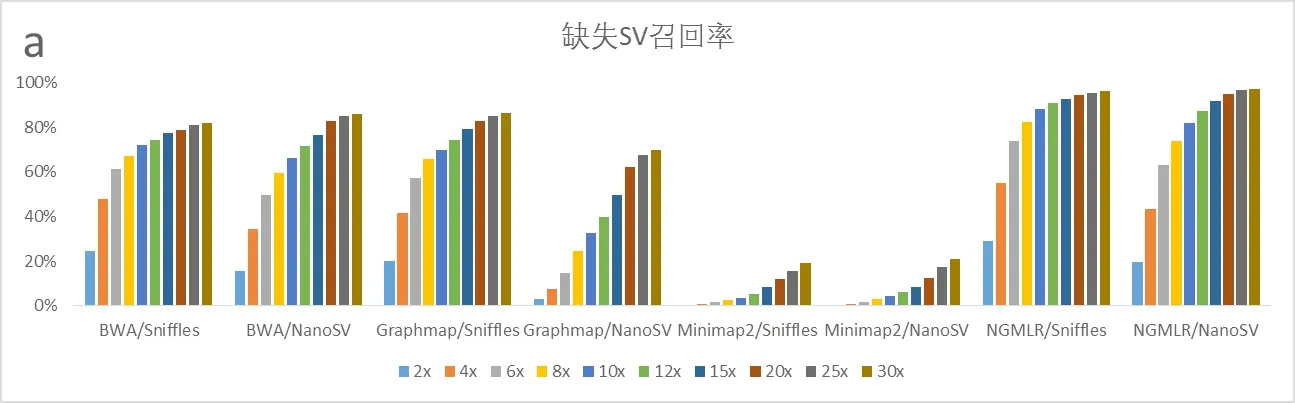
图3 各流程覆盖度下对于缺失SV 的召回率
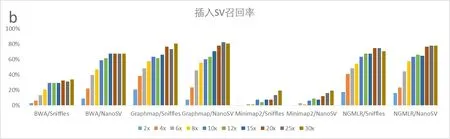
图4 各流程各覆盖度的召回率
3 讨论
在本研究中,我们评估了目前常用于Naopore 测序数据分析的4 种比对软件。以及两种SV 发现软件。我们发现对于nanopore 测序来说,20×的覆盖度是在研究中比较适合的覆盖度。同时,我们发现不同的分析流程之间结果会有很大的差异,对于缺失SV,20×覆盖度下,召回率最高为NGMLR/NanoSV(召回率:94.918%,准确率:2.463%);对于插入SV,20×覆盖度下,召回率最高为Graphmap/NanoSV(召回率:77.941%,准确率:0.122%)
在发现SV 的数量上,Sniffles 与NanoSV 相比总体上差异不大,但Sniffles 可以发现复杂结构变异这一点上更具优势。
猜你喜欢
科学技术创新(2022年30期)2022-10-21
军事文摘(2022年16期)2022-08-24
农业与技术(2021年23期)2021-12-14
农业与技术(2021年16期)2021-08-31
今日农业(2021年11期)2021-08-13
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
支部建设(2020年15期)2020-07-08
西安科技大学学报(社会科学版)(2020年1期)2020-04-01
百科知识(2015年18期)2015-09-10
