
优化大肠杆菌CRISPR/Cas9基因编辑系统及应用
2020-08-19 02:51邵梦瑶路福平朱欣娜张学礼
生物学杂志 2020年4期
邵梦瑶, 路福平, 朱欣娜, 张学礼
(1. 天津科技大学 生物工程学院, 天津 310018; 2. 中国科学院 天津工业生物技术研究所, 天津 300308; 3. 中国科学院 系统微生物工程重点实验室, 天津 300308)
大肠杆菌被广泛应用于发酵工程和代谢工程等诸多生物学领域[1]。为使大肠杆菌成为生产有机酸[2]和氨基酸[3-4]等重要的工程菌株,敲除及整合外源功能基因是常常用到的策略。通常的技术手段是利用Red重组酶进行同源重组实现基因的敲除和整合,但该技术方法操作繁琐。因此,研究人员对简单快速的基因编辑系统的需求越来越迫切。
2012年,Jennifer A. Doudna[5]阐明了Type II CRISPR系统中的Cas9核酸酶(来源于化脓链球菌Streptococcuspyogenes)的功能,这个160 ku的核酸酶结合两个RNA(crRNA和tracrRNA)就可以实现对双链DNA的切割。这为新的基因编辑系统奠定了最根本的基础。
2013年,张峰课题组[6]证实了CRISPR/Cas9基因编辑系统能够实现对原核微生物如链球菌和大肠杆菌的基因编辑。该研究虽然证明了CRISPR/Cas9[7]能对大肠杆菌进行基因编辑,但并没有建立起良好的可操作体系。2015年,美国伊利诺伊大学香槟分校赵惠民课题组[8]和中科院上海植生所杨晟课题组[9]分别在链球菌和在大肠杆菌中建立了完善的 CRISPR/Cas9 基因编辑系统。这两个系统都将crRNA和tracrRNA融合表达出gRNA,不同之处前者是单质粒编辑系统,后者是双质粒编辑系统。单质粒编辑系统[10]将Cas9,gRNA和供体DNA构建在同一个表达载体上,在遗传操作过程中,只需一轮质粒转化就能实现对基因组的编辑。但单质粒由于本身骨架片段较大,可插入的供体DNA大小受限,质粒构建会有一定的难度。双质粒编辑系统可以解决单质粒构建困难的问题,将Cas9,Red构建在一个质粒上,将目标N20-gRNA, 供体DNA片段构建在另一个质粒上,利用这个双质粒CRISPR/Cas9系统,大肠杆菌单基因编辑的效率达到100%。
本实验室在2016年建立了CRISRP/Cas9单质粒基因编辑系统,单基因编辑效率达到100%。随后,构建了双质粒基因编辑系统[11],实现了多个严谨型启动子的高效整合(编辑效率达到100%),为快速评价启动子在染色体上的功能提供了可能。然而,将该系统用于于工程菌株的构建时,我们发现,在连续进行基因的敲除[12]或外源基因的整合过程中,供体质粒很难通过传代培养及电击[16]的方式消除,影响了工程菌株的构建进程。
因此,本文旨在构建拥有自剪切功能的供体质粒,优化已有的CRISPR/Cas9基因编辑系统,并应用于衣康酸工程菌株的构建中,实现了基因的连续敲除和整合,快速地得到工程菌株,为工程菌株的构建和改造提供一种更为便捷的高效方法。
1 材料与方法
1.1 菌种来源
本实验所用菌株和质粒见表1。

表1 实验所用的菌株和质粒
1.2 培养基和培养条件
LB培养基[14],用于大肠杆菌的培养,37 ℃,250 r/min。NBS培养基[18],用于衣康酸的发酵,30 ℃,200 r/min。
1.3 试剂及仪器
抗生素的使用浓度分别为:氨苄霉素100 μg/mL,卡那霉素50 μg/mL,氯霉素34 μg/mL。诱导剂:0.5 mmol/L IPTG和1% L(+)阿拉伯糖。抗生素和诱导剂,美国SIGMA-ALDRICH试剂公司;PCR高保真扩增Phusion酶,快速T4 DNA连接酶,限制性内切酶BsaI,美国NEB生物公司;Trans T1 感受态细胞,中国全式金生物技术有限公司;Sigma 3-18K离心机,德国SIGMA实验室离心机公司;全自动凝胶成像仪,美国Bio-rad生物公司。
1.4 含有自剪切元件的供体质粒pV4构建
以placZ质粒为模板,使用引物Bone-F和Bone-R进行反向PCR扩增得到PCR产物I,用引物cat-N20-up和cat-N20-down进行PCR扩增,得到PCR产物II;以pACYC184-M质粒[14]为模板,用引物lacI-Ptrc-up和lacI-Ptrc-down进行PCR扩增,得到PCR产物III。PCR产物用DpnI处理模板DNA,用Golden Gate 技术组装策略[15]将PCR片段I、II和III进行组装,得到供体pV4质粒。质粒见表1,引物序列见表2。
1.5 CRISPR/Cas9基因编辑系统中自剪切供体质粒的构建
1.5.1 敲除型供体质粒pV4-del-gene的构建
以pV4质粒为模板,用引物对N20-B-F1/N20-B-R1扩增获得pV4骨架片段,用引物gene-N20-B-F2/N20-B-R2扩增出针对目标基因gene-N20-gRNA的片段;以E.coliATCC 8739 DNA为模板,用引物gene-F1/gene-R1和gene-F2/gene-R2分别PCR扩增得到敲除基因的上下游同源臂序列。用材料和方法1.4中的组装策略得到pV4-del- gene质粒。该通用方法构建pV4-del-ptsI和pV4-del-iclR供体质粒,引物命名将gene替换为ptsI或iclR(表2)。

表2 本研究所用的引物及调控元件序列
1.5.2 整合型供体质粒pV4-ins-gene的构建
和构建敲除型质粒方法1.5.1类似,除了扩增pV4骨架片段、针对目标基因的gene-N20-gRNA片段和基因的上下游同源臂片段外,还要扩增需要整合的基因片段。
以pUC57-kan-M1-93-cad质粒为模板,用引物93-cad-F-GGTG和93-cad-R-CTGG扩增出大小1.2 kb的片段。该片段和pV4骨架片段,araBAD-N20-gRNA片段,araBAD基因上下游同源臂片段进行组装和转化,得到pV4-del-araBAD-ins-cad质粒。
1.6 利用CRISPR/Cas9基因编辑系统进行基因的敲除和整合
具体过程见文献[11]。
1.7 CRISPR/Cas9编辑系统中pV4系列供体质粒的消除
当CRISPR/Cas9双质粒(pRedCas9和pV4系列供体质粒)编辑系统完成某个基因的敲除或整合后,将PCR验证正确的单克隆在LB (含0.1 mmol/L IPTG)平板上划线,37 ℃培养,10~12 h得到单克隆。将单克隆分别点板于LB氯霉素和LB平板,37 ℃培养3~5 h。观察氯霉素平板上不长,LB平板上生长的单克隆,即为消除pV4系列供体质粒的菌株。
2 结果和分析
2.1 大肠杆菌CRISPR/Cas9基因编辑系统优化
2.1.1 大肠杆菌CRISPR/Cas9基因编辑系统优化原理及使用范围
在大肠杆菌中构建代谢途径时,常常需要连续的进行基因敲除或者整合。利用现有的CRISPR/Cas9双质粒系统进行基因的编辑时,供体DNA质粒的消除是实现基因连续编辑的前提。虽然采用连续的划线传代及电击的方法可以消除供体DNA质粒[16],但是耗时长、消耗大量的人力和物力。为提升代谢工程菌株构建的效率,有必要对已有的CRISPR/Cas9双质粒基因编辑系统进行优化,提高供体DNA质粒消除效率。因此,本研究对前期的CRISPR/Cas9双质粒基因编辑系统进行了思考,优化了该编辑系统中的供体DNA质粒(图1),在供体DNA质粒上添加了自剪切功能元件,使完成基因编辑后,在Cas9蛋白和IPTG的诱导下实现供体DNA质粒的自剪切,从而方便下一轮基因的编辑。
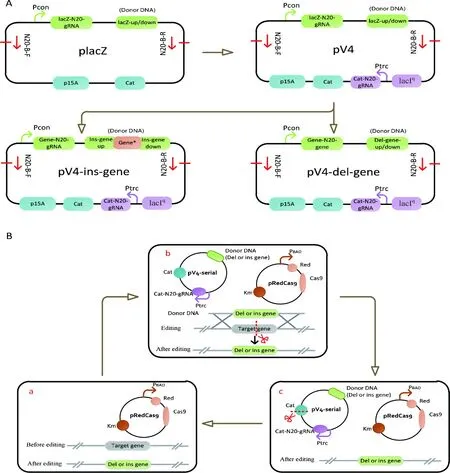
A:优化CRISPR/Cas9编辑系统供体DNA质粒。 B:优化CRISPR/Cas9编辑系统,实现基因的连续编辑。a:在大肠杆菌中转化pRedCas9质粒;b:转化含有供体DNA且具有自剪切功能的pV4质粒,对目标基因进行编辑(敲除或者整合);c:编辑后,在Cas9和IPTG的诱导下实现供体DNA质粒自剪切消除,从而可以进行下一轮的基因编辑。骨架片段通用引物对N20-B-F/N20-B-R;Pcon: 组成型启动子;lacIq调控Ptrc启动子;cat氯霉素基因,P15A复制子;Red重组;Cas9蛋白;PBAD 为组成型启动子
供体DNA质粒具体的优化改造过程如图1-A所示。从实验室已有的供体DNA质粒placZ出发,在其质粒的骨架区域上添加了针对氯霉素抗性基因(cat)的N20-gRNA序列元件,该元件在Ptrc启动子下表达。在IPTG的诱导下,Ptrc启动子表达,转录得到cat-N20-gRNA,该前导gRNA识别并与cat基因的特异20 bp结合,在Cas9蛋白的帮助下实现对供体DNA质粒pV4的剪切。当把敲除或整合基因片段连接于pV4质粒的骨架片段上(图1-A),就能获得敲除型或整合型自剪切pV4供体质粒(pV4-del-gene或pV4-ins-gene)。图1-B示意了利用自剪切供体pV4系列质粒的CRISPR/Cas9编辑系统,实现对基因连续编辑的过程。
2.1.2 自剪切pV4质粒作为供体质粒的优势
我们构建的自剪切供体质粒,可以在IPTG的诱导下及Cas9蛋白的帮助下实现对供体DNA质粒pV4的剪切。与没有针对氯霉素抗性基因(cat)的N20-gRNA序列的pV4质粒相比,它的质粒消除时间短,优化实验室的CRISPR/Cas9基因编辑系统,实现基因的连续敲除或整合,节约了基因操作的时间,有广泛的工程菌株构建应用前景。
2.2 优化CRISPR/Cas9基因编辑系统中的供体质粒
为优化双质粒CRISPR/Cas9基因编辑系统,实现对基因的连续编辑,对供体DNA质粒进行了改造:在供体质粒placZ的骨架区域添加上3个元件:cat-N20-gRNA元件,以placZ质粒为模板,用引物cat-N20-up/cat-N20-down扩增得到,大小400 bp(图2-A,泳道2);元件lacIq和元件Ptrc在质粒pMACYC184上相邻,用引物lacI-Ptrc-up/lacI-Ptrc-down扩增得到,大小1.5 kb(图2-A,泳道3)。将这3个元件和placZ反向扩增的骨架片段(图2-A,泳道1)用Golden Gate策略组装,得到pV4质粒(图2-B)。pV4质粒含有自剪切元件lacIq-Ptrc-cat-N20-gRNA,在IPTG诱导和Cas9蛋白作用下,可以实现质粒的自我消除。

A: pV4质粒组装PCR片段; B: pV4质粒; M: DNA marker trans 2K plus
2.3 应用优化的CRISPR/Cas9基因编辑系统快速构建工程菌株
为显示优化后的CRISPR/Cas9 基因编辑系统的方便快捷,将其应用于大肠杆菌产衣康酸工程菌株的构建。构建大肠杆菌衣康酸工程菌,需要敲除ptsI(糖磷酸转移酶系统酶I)基因,iclR(异柠檬酸裂解酶调节剂)基因及整合cad(顺乌头酸脱羧酶)基因。
2.3.1 构建敲除型pV4-del-gene和整合型pV4-ins-gene质粒
为敲除ptsI、iclR基因和整合cad基因,首先要构建两个敲除型质粒pV4-del-ptsI和pV4-del-iclR,一个整合型质粒pV4-del-araBAD-ins-cad,将cad基因整合到大肠杆菌araBAD基因位点。
以pV4质粒为DNA模板,扩增得到pV4系列质粒的通用骨架片段,大小为4.4 kb(图3-A、B、C,泳道1),含有氯霉素抗性基因cat和复制子P15A序列及自剪切元件lacI-Ptrc-cat-N20-gRNA。同时,以pV4质粒为模板,得到400 bp的针对目标基因的N20-gRNA,分别为ptsI-N20-gRNA,iclR-N20-gRNA及araBAD-N20-gRNA (图3-A、B、C,泳道2)。以ATCC 8739 DNA为模板,扩增得到ptsI、iclR上下游400 bp(图3-A、B,泳道3和4)和araBAD上、下游约400~500 bp的DNA片段(图3-C,泳道3和5)。以pUC57-kan-M1-93-cad质粒DNA模板, 扩增得到cad基因和M1-93启动子序列,大小约1 kb (图3-C,泳道4)。
按照Golden Gate的片段组装策略,分别组装ptsI、iclR敲除型质粒和cad整合型质粒。组装产物转化Trans T1感受态细胞,得到的克隆PCR验证分析后送样测序,组装正确的效率达到100%。pV4-del-ptsI,pV4-del-iclR和pV4-del-araBAD-ins-cad的质粒电泳图,见图3-D 泳道1、2和3。
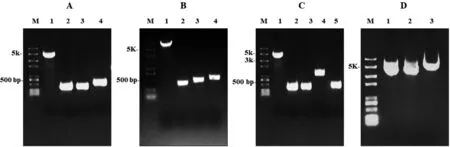
A:组装pV4-del-ptsI质粒的PCR片段; B:组装pV4-del-iclR质粒的CR片段; C:组装pV4-del-araBAD-ins-cad质粒的PCR片段; D:供体pV4系列质粒。M: DNA marker trans 2K plus
2.3.2 基因的连续敲除及其供体质粒的消除
从野生型大肠杆菌E.coliATCC 8739出发,依次敲除ptsI基因和iclR基因,整合cad基因。
将含有pRedCas9和供体质粒的pV4-del-ptsI的大肠杆菌,接种于LB液体培养基(含阿拉伯糖),诱导同源重组Red敲除ptsI基因和表达Cas9蛋白切割未发生同源重组的染色体。在含有阿拉伯糖的LB平板上划线,得到单克隆,用ptsI-YZ-up/ptsI-YZ-down引物进行ptsI基因敲除验证。结果显示:验证的6个克隆ptsI基因均被敲除,其电泳条带大小为750 bp(图4-A左,泳道1~6),对照未敲除的大小是2 kb(图4-A左,泳道7) 。
供体质粒pV4-del-ptsI的消除:将验证正确的克隆在LB(含0.1 mmol/L IPTG,50 μg/L 卡纳霉素)平板上划线,在IPTG的诱导和得到单克隆。将单克隆分别点板于氯霉素和LB平板。氯霉素平板上不长,LB平板上生长的单克隆,即为消除pV4-del-ptsI供体质粒的菌株(图3-A右),命名为YM-I001,质粒消除效率达到100%(图4-A右,表3)。
从YM-I001出发,按照敲除ptsI基因的方式,继续敲除iclR基因。PCR验证(用iclR-YZ-up/iclR-YZ-down引物)4个克隆,显示iclR基因均被敲除,其电泳条带大小为750 bp(图4-B左,泳道1~4),未被敲除的大小是2 kb(图4-B左,泳道5)。按照消除pV4供体质粒的方式消除pV4-del-iclR,消除效率达到98%(图4-B右,表3),消除供体质粒pV4-del-iclR的菌株命名为YM-I002。
从YM-I002出发,利用pV4-del-araBAD-ins-cad为的供体质粒引入cad基因。整合后,用araBAD-YZ-up/cad-YZ-down200引物进行PCR验证,cad基因整合了的电泳条带大小为1.7 kb(图4-C左,泳道1~7),未被整合的对照无条带(图4-C左,泳道8)。按照消除pV4供体质粒的方式消除pV4-del-araBAD-ins-cad,消除效率达到94%(图4-C右,表3),相应的菌株命名为YM-I003,该菌株能产衣康酸1.5 g/L。

A:敲除ptsI基因及其供体质粒的消除; B:敲除iclR基因及其供体质粒的消除; C:整合cad基因及其供体质粒的消除; M: DNA marker trans 2K plus

表3 供体质粒消除效率及其消耗时间
3 小结
本研究优化了双质粒CRISPR/Cas9编辑系统中的供体质粒,优化后的pV4系列供体质粒不仅能在基因编辑过程中提供针对同源重组的供体DNA片段和提供针对目标基因的N20-gRNA,而且能够在基因编辑结束后,通过表达cat-N20-gRNA实现供体质粒的自我消除,方便下一轮供体质粒进入实现目标基因的编辑。
将优化后的双质粒CRISPR/Cas9编辑系统,应用于大肠杆菌产衣康酸工程菌株的构建,实现了基因的连续编辑,成功敲除了ptsI基因、iclR基因,在araBAD基因位点整合了cad基因。衣康酸工程菌株的构建在3周内完成,该菌株的快速构建,得益于编辑过程中pV4系列供体质粒的自我剪切,pV4系列供体质粒的消除过程需要的时间总计13~17 h(表3)。
本研究结果为大肠杆菌工程菌株的构建提供了更为优化的工具,提高了双质粒CRISPR/Cas9编辑系统连续进行基因编辑的效率,有广阔的应用前景。
猜你喜欢
中国医药导报(2021年35期)2022-01-20
矿产勘查(2020年11期)2020-12-25
数学大王·低年级(2020年8期)2020-08-14
航空发动机(2020年3期)2020-07-24
华南师范大学学报(自然科学版)(2020年3期)2020-07-01
首都医科大学学报(2019年5期)2019-10-24
生物工程学报(2019年1期)2019-01-30
小雪花·初中高分作文(2016年9期)2016-05-14
铁道科学与工程学报(2015年4期)2015-12-24
郑州大学学报(医学版)(2015年2期)2015-02-27
