
位阻和电子效应对位阻型二芳基乙烯光响应性能的影响
2020-11-06 08:32李萌祺张志鹏朱为宏
华东理工大学学报(自然科学版) 2020年5期
李萌祺, 张志鹏, 朱为宏
(华东理工大学化学与分子工程学院,精细化工研究所,上海 200237)
光致变色染料[1-3]可通过外加光照对化合物的物理和化学性质进行可逆调控,越来越被人们所重视,在光信息存储[4-5]、自组装[6-9]、液晶[10-11]和荧光成像[12-13]等领域有着广泛的应用。在众多光致变色染料中,二芳基乙烯化合物[4,14]由于其优异的高双稳态[15]和高抗疲劳度[16]等优点,受到格外关注。人们也通过对烯桥和侧端的一系列修饰来进一步提升二芳基乙烯化合物的光响应性能。一般而言,烯桥相比于侧端更接近二芳基乙烯的核心区域,通过对烯桥的修饰,往往能够带来更多光照前后性能上的差异。研究人员通过引入多种新型烯桥(如噻咯[17]、香豆素[18]、苯并噻吩砜[19]、卟啉[20]和苯并二噻二唑[21-22]等)对二芳基乙烯体系进行了修饰,其中引入低芳香性的烯桥[23]体系往往能有效降低闭环体能级,使之具有更为优异的双稳态性能,这在二芳基乙烯体系的实际应用中具有重要意义。
一般来说,二芳基乙烯开关态(Open Form, o-form)原是无色的,当受到光照后会产生有色的闭环态(Closed Form, c-form)。进一步从结构上细分,二芳基乙烯开环态具有两种构象异构体,一种是具有光响应性的反平行构象异构体(Anti-parallel Conformer,ap-conformer),另一种是光惰性的平行构象异构体(Parallel Conformer, p-conformer), 根 据Woodward-Hoffmann 规则,只有反平行异构体才能发生光致电环化反应生成闭环态。一般而言,由于传统二芳基乙烯体系的侧端基团位阻较小,两种构象异构体能在溶液中发生快速交换,不能分离,这使得二芳基乙烯的闭环量子效率往往低于50% (摩尔分数),因此增加反平行异构体的比例是提高闭环量子效率最直接的办法[24-25]。本课题组之前报道了一种低芳香性的位阻型苯并二噻二唑烯桥,并通过引入大位阻侧端苯并噻吩基团,彻底阻断常规条件下平行与反平行异构体间的转化,使之具有较高的量子效率和双稳态性能[21,26]。
因此,基于上述研究,本文系统地比较了侧端位阻效应,并通过引入给电子的对甲氧基苯基和吸电子的吡啶基团,分析了位阻型二芳基乙烯体系光响应性能的影响。
1 实验部分
1.1 原料和试剂
无水碳酸钠,w=99%,上海泰坦科技股份有限公司;无水碳酸钾,w=99%,上海泰坦科技股份有限公司;四氢呋喃(THF),w=99%,上海泰坦科技股份有限公司;4-溴吡啶盐酸盐,w=99%,阿达玛斯试剂有限公司;正丁基锂(n-BuLi),2.5 mol/dm3正己烷溶液,北京百灵威科技有限公司;硼酸三甲酯(B(OMe3)),w=99%,阿达玛斯试剂有限公司;四(三苯基膦)钯(Pd(PPh3)4),w=99%,北京百灵威科技有限公司;THF 使用前进行干燥处理,常压蒸馏后方可使用。
1.2 测试与表征
核磁共振氢谱和碳谱的测定采用了德国Bruker 公司的Bruker Avance 超导傅里叶变换核磁共振波谱仪(400 MHz),室温下以四甲基硅烷(TMS,摩尔分数0.03%)为内标测定,氘代氯仿(CDCl3, δ =7.26)、氘 代 苯(C6D6, δ = 7.16)和 氘 代 四 氢 呋 喃(Tetrahydrofuran-d8, δ = 1.73 和3.58)为溶剂;质谱采用的测试仪器为美国沃特世公司的Waters LCT Premier XE spectrometer;紫外-可见光吸收光谱采用美国安捷伦公司的Agilent Cary 60(静态光谱)和在线光学平台(青岛OceanOptics,动态光谱)进行测定。光致变色反应由装有适当波长窄带干涉滤光片(沈阳汇博光学技术)的 λ = (313 ± 10) nm 的Hg/Xe灯(Hamamatsu, LC8 Lightingcure, 200 W)照射引发。光强由美国Ophir 公司的Ophir (PD 300-UV)光电二极管测定。荧光由日本Horiba 公司的Floromax-4 Spectroflorometer 检测。
1.3 目标化合物1 和2 的合成
目标化合物1~4 的平行与反平行异构体和闭环体的结构式见表1。其中2a 和2p 以及4a 和4p 无法分离,表达为2o 和4o。目标分子1a 和1p 以及2o 和2c 的合成路线见图1。
目标化合物3[26]和4[23]的合成参考之前的工作。化合物5 与4-溴吡啶盐酸盐经Suzuki 偶联反应,得到中间体6,而后正丁基锂进攻化合物6 的4 号位溴原子制备得到硼酸后,不经处理直接与苯并二噻二唑烯桥发生偶联得到目标化合物1a 和1p。目标化合物2 则直接由中间体8 与烯桥经Suzuki 偶联反应得到目标化合物2o。

表 1 目标化合物1~4 的开环体的平行与反平行异构体和闭环体的化学结构式Table 1 Chemical structures of parallel and anti-parallel conformer of open form as well as closed form of the target compounds 1~4
1.3.1 目标化合物1 的合成 中间体6 的合成:于500 mL 三口瓶中,依次加入化合物5[26](4.78 g,20.32 mmol)、碳酸钠(25.44 g,0.24 mol)、四氢呋喃(240 mL)、水 (120 mL)、溴代吡啶盐酸盐(9.88 g,50.8 mmol)和四(三苯基膦)钯 (0.46 g, 0.40 mmol)作为催化剂,在氮气保护加热至85 ℃反应8 h,反应液由黄色变为蓝色再变为墨绿色。反应结束后,分液去水相,将有机相旋转蒸发浓缩,加入氯化钠水溶液(150 mL),采用乙酸乙酯(2 × 100 mL)萃取,旋转蒸发仪蒸去溶剂后进行柱层析分离 (二氯甲烷与甲醇体积比为 150∶1),并且每300 mL 滴加一滴三乙胺防止拖尾。展开剂极性梯度增加,每300 mL 补加0.5 mL甲醇和1 滴三乙胺。收集第2 个组分,得到黄色固体,用甲醇重结晶得到淡黄色固体4.17 g (产率49.5%,摩 尔 分 数)。1H-NMR (400 MHz, CDCl3, δ): 2.33 (s,3H, −CH3), 2.45 (s, 3H, −CH3), 7.31 (d, J = 6.0 Hz, 2H,pyridine-H), 8.62 (d, J = 6.0 Hz, 2H, pyridine-H)。13CNMR (100 MHz, CDCl3, δ): 15.37, 15.89, 114.63,122.92, 131.56, 134.55, 134.66, 142.09, 150.12。

图 1 目标分子1a 和1p 以及2o 和2c 的合成路线Fig. 1 Synthetic route of target molecules 1a and 1p as well as 2o and 2c
中间体7 的合成:于250 mL 干燥过的三口烧瓶中,加入反应物9 (4.17 g,15.56 mmol)和四氢呋喃(100 mL),于氮气保护下低温搅拌,用注射器滴加2.5 mol/dm3正丁基 锂(6.79 mL,16.99 mmol),反应45 min 后加入硼酸三甲酯(2.12 mL,18.59 mmol),关闭制冷机,搅拌1 h 后升至室温搅拌过夜。然后于100 mL 单口烧瓶中旋转蒸发仪蒸去溶剂后直接投入下一步反应。
目标化合物1a 和1p 的合成:于100 mL 单口烧瓶中,用旋转蒸发仪蒸去上次反应液,直接加入原料二溴苯并二噻二唑(0.684 g,1.945 mmol)、碳酸钾(6.9 g,0.05 mol)、二氧六环 (50 mL)、水(25 mL)和四(三苯基膦)钯(400 mg,0.346 mmol),在氮气保护下避光加热至130 ℃反应回流20 h 后停止反应,薄层层析分离(展开剂二氯甲烷和甲醇的体积比为10∶1),第2 个点为变色点即反平行构象,而第3 点为平行构象。加水用二氯甲烷萃取后旋干溶剂柱色谱分离(二氯甲烷与甲醇的体积比为150∶1),随后不断增大极性,每300 mL 加2 mL 甲醇,依次不断增大,收集第二、第三组分,分别得到反平行与平行异构体粗品,用乙酸乙酯重结晶,得浅黄色反平行异构体351 mg(产率31.8%,摩尔分数)和黄色平行异构体202 mg(产率18.3%,摩尔分数)。
1a:1H-NMR (400 MHz, CDCl3, δ): 2.04 (s, 6H,−CH3), 2.08 (s, 6H, −CH3), 7.37 (d, J = 6.0 Hz, 4H,pyridine-H), 8.62 (d, J = 6.0 Hz, 4 H, pyridine-H)。13CNMR (100 MHz, CDCl3, δ): 14.97, 16.00, 122.99,132.03, 132.63, 134.25, 135.17, 138.94, 142,16, 147.44,150.07, 156.88。HRMS (TOF MS ES+ for [M+H]+):calcd. for C28H21N6S4: 569.071 1; found 569.070 7。
1p:1H-NMR (400 MHz, CDCl3, δ): 1.96 (s, 6H,−CH3), 2.17 (s, 6H, −CH3), 7.34 (d, J = 5.6 Hz, 4H,pyridine-H), 8.62 (d, J = 5.6 Hz, 4H, pyridine-H)。13CNMR (100 MHz, CDCl3, δ): 15.37, 15.76, 122.98,131.86, 132.60, 134.42, 135.11, 139.49, 142.19, 147.42,150.08, 156.85。HRMS (TOF MS ES+ for [M+H]+):calcd. for C28H21N6S4: 569.071 1; found 569.070 4。
1.3.2 目标化合物2o 和2c 的合成 在200 mL 单口瓶中,依次加入化合物8[27](15.8 mmol)、苯并二噻二唑烯桥(1.1 g, 3.12 mmol)、四(三苯基膦)钯(200 mg,0.17 mmol)、二氧六环(100 mL)、碳酸钾(9.5 g,0.1 mol)的水(50 mL)溶液,氮气保护回流(130 ℃)反应20 h。冷却至室温后加入水(70 mL),用二氯甲烷萃取,浓缩后以二氯甲烷与甲醇(二氯甲烷与甲醇的体积比为100∶1)为展开剂进行柱层析,得到黄褐色固体,乙酸乙酯重结晶,过滤,滤饼用乙酸乙酯(30 mL)洗涤3 次,真空干燥,得700 mg 白色粉末。滤液稀释至250 mL,紫外光照10 h 后,浓缩,以二氯甲烷与甲醇(二氯甲烷与甲醇体积比为300∶1)为展开剂进行柱层析,得150 mg 闭环体,开环体和闭环体共得850 mg(产率50.3 %,摩尔分数)。1H-NMR (400 MHz, CDCl3,δ): 2.15 (s, 3H, −CH3, ap-conformer), 2.26 (s, 3H,−CH3, p-conformer), 7.24 (s, 1H, thiophene-H, apconformer), 7.31 (d, 2H, J = 6.0 Hz, pyridine-m H, apconformer), 7.39 (d, 2H, J = 6.0 Hz, pyridine-m H, pconformer), 7.47 (s, 1H, thiophene-H, p-conformer), 8.54(d, 2H, J = 6.0 Hz, pyridine-o H, ap-conformer), 8.58 (d,2H, J = 6.0 Hz, pyridine-o H, p-conformer)。13CNMR(100 MHz, CDCl3, δ): 15.12, 15.16, 119.49, 127.76,127.79, 130.23, 130.39, 133.08, 133.15, 137.41, 137.54,140.71, 140.88, 140.92, 141.51, 147.44, 150.29, 156.41,156.70。HRMS (TOF MS ES+ for [M+H]+: calcd. for C26H17N6S4: 541.039 8; found 541.039 5。2c:1H-NMR(100 MHz, CDCl3, δ): 2.36 (s, 6H, −CH3), 7.52 (d, 4H,J = 6.0 Hz, pyridine-m H), 8.42 (s, 2H, thiophene-H),8.72 (d, 4H, J = 6.0 Hz, pyridine-o H)。
2 结果与讨论
2.1 目标分子的设计
长期以来,传统二芳基乙烯体系侧端芳基较小的位阻使得侧端芳基能快速旋转,导致平行与反平行构象异构体发生快速交换而无法分离。而由于平行异构体不具有光响应性,导致闭环量子效率长期限制在50% (摩尔分数)以下。之前,本课题组报道了独特的位阻型苯并二噻二唑烯桥体系,通过引入大位阻的侧端芳基,能成功分离平行与反平行异构体,因此有效突破了闭环量子效率50% (摩尔分数)的限制[21,26]。本文中,基于课题组之前的研究,合成了目标化合物1 和2,目标分子1 在噻吩β 位引入大位阻取代基,期望同样分离平行与反平行两种构型。同时比较β 位取代噻吩体系1 和3 与小位阻的未取代噻吩体系2 和4 之间的光致变色性能,并且也进一步比较侧端引入吸电子的吡啶基团和供电子的对甲氧基苯基基团对于位阻型二芳基乙烯体系光致变色性能的影响。
2.2 目标分子的核磁氢谱解析
在得到了4 个目标化合物后,对它们进行核磁表征。核磁氢谱可以有效区分二芳基乙烯的多重异构体,同时也是研究光致变色反应的有效工具[28]。

图 2 目标化合物1 在C6D6 中的1H-NMR 谱(400 MHz, 293 K)Fig. 2 1H-NMR spectra (400 MHz, 293 K) of target compound 1 in C6D6
首先对目标化合物1 的核磁氢谱进行解析,如图2所示(为了显示更为清晰,低场区域(阴影部分)信号峰放大了两倍,图中虚线表示不同化合物核磁出峰位移变化)。由于β 位取代噻吩引入的甲基与烯桥的距离非常近,这样可产生一定的位阻效应,使得目标化合物1 能够实现平行与反平行异构体的分离。实际上,1a 和1p 相互之间无法转化,因此可以通过传统柱层析进行分离。两者在高场都显示了两组甲基峰,其中处于较低场的甲基峰为噻吩2 号位甲基峰(1a: δ = 2.06, 1p: δ = 2.15),而较高场的甲基峰则为β 位取代甲基(1a: δ = 2.03, 1p: δ = 1.95)。相比于平行异构体,反平行异构体的2 号位甲基处于相对高场,这是由于反平行异构体中2 号位甲基位于另一个噻吩环平面的正上方,受到噻吩环电流的屏蔽作用所导致的,该现象可以有效区分平行与反平行异构体。另一方面,β 位取代甲基出现在更为高场的区域,是由于受到烯桥环平面所带来的明显的屏蔽作用,说明甲基与烯桥平面之间有显著的空间效应。低场则相应地显示两组吡啶上的氢信号峰,两个异构体间差异不大。对1a 进行紫外光照(λ = 313 ± 10 nm),则能相应地生成闭环体1c 的信号峰。
目标化合物2 不具有β 位取代甲基,因此无法分离得到平行与反平行异构体。从核磁氢谱中可以同时观察到平行与反平行异构体两组信号峰,如图3所示(虚线表示不同化合物核磁出峰位移变化)。其中高场可以发现两组甲基峰,分别是平行与反平行两个构象异构体的甲基峰(反平行构象:δ = 2.20;平行构象:δ = 2.27)。当受到紫外光照后,原本两组信号峰逐渐变为一组信号峰,对应于闭环体2c,这也充分说明了平行与反平行异构体可以互相转化。值得一提的是,氢原子b (Hb)的信号峰相比于开环体明显偏向了低场,这是由于Hb与烯桥噻二唑氮原子间形成分子内氢键的作用[21,26]。以上结果充分说明了β 位取代甲基可以有效增加侧端与烯桥之间的位阻,从而阻断平行与反平行构型间的转化,实现两者的彻底分离。
相比于上述两个化合物,目标化合物3 和4 之间也呈现出类似的结果,目标化合物3 由于β 位取代甲基的影响,构象异构体3a 和3p 可以分离。图4 示出了目标化合物3 在氘代四氢呋喃中的核磁氢谱(虚线表示不同化合物核磁出峰位移变化)。图4 中核磁氢谱高场显示了两组甲基峰,分别为α 位甲基和β 位取代甲基,其分布与目标化合物1 基本一致。而在低场,两者之间的信号峰差异较小。同样当受到紫外光照(λ = (313 ± 10) nm)后,3a 发生光致变色反应,显示出相应的3c 的信号峰。目标化合物4 则显示了较弱的空间位阻效应,无法分离平行与反平行异构体。从核磁氢谱图上可以同时观察到目标化合物4o 两个构象异构体的信号峰,且当受到紫外光照后,两组信号峰会逐渐消失并只产生一组新的信号峰,说明了两个异构体可以互相转化。同样的,在4c 的谱图上能够发现由于分子内氢键所导致的Hb信号峰向低场移动的现象,如图5 所示(为了显示更清晰,低场区域(阴影部分)信号峰放大了两倍,图中虚线表示不同化合物核磁出峰位移变化)。
2.3 目标分子1~4 的吸收和荧光性能

图 3 目标化合物2 在tetrahydrofuran-d8 中的1H-NMR 谱(400 MHz, 293 K)Fig. 3 1H-NMR spectra (400 MHz, 293 K) of compound 2 in tetrahydrofuran-d8

图 5 目标化合物4 在氘代四氢呋喃中的1H-NMR 谱(400 MHz, 293 K)Fig. 5 1H-NMR spectra (400 MHz, 293 K) of compound 4 in tetrahydrofuran -d8
2.3.1 反平行异构体或开环体的光致变色性能 首先对目标化合物1~4 的反平行异构体的光致变色性能进行测试。由于目标分子2 和4 的反平行与平行异构体无法分离,因此统一讨论它们开环体即2o 和4o 的光致变色性能。1a、2o、3a 和4o 在四氢呋喃溶液中都只在200~400 nm 紫外区有吸收,因此呈现为无色透明溶液。4 种化合物均在290 nm 左右具有强吸收带,同时在350~430 nm 左右也都有中等强度的肩峰,这是由于烯桥与侧端之间分子内电荷转移(Intramolecular Charge Transfer, ICT)所产生的。当4 种化合物的四氢呋喃溶液经过(313 ± 10)nm 的紫外光照后,其吸收光谱发生了明显改变,在可见光区域出现了新的吸收峰,这是由于典型的光致变色电环化反应所导致的,所产生的闭环体的共轭体系面积更大,因此在可见光区会产生新的吸收,也导致1a、2o、3a 和4o 溶液的颜色由原来的无色分别变为红棕色、绿色、紫红色和墨绿色,分别对应各自闭环态的最大吸收波长,即561、649、569 nm 和654 nm。4 种化合物达到光稳态(Photostationary State, PSS)的转化率都超过97%(摩尔分数,见图6,图6 中箭头朝上表示峰的强度增强,箭头向下表示峰的强度减弱)。6(a)、6(c)中1c 和3c 的吸收和荧光由谱图计算得到。
值得一提的是,相比于小位阻体系2 和4,大位阻目标分子1 和3 闭环后最大吸收波长要明显偏短。1c 的最大吸收波长相比于2c 蓝移了约88 nm,同时1c 的光谱相较于2c,也缺失了许多精细结构,例如在413 nm 处的强吸收带,1c 明显不具有2c 中左侧的肩峰。对于3c 和4c 的吸收光谱也能得出类似的结论。说明β 位取代甲基的巨大位阻使得闭环反应发生时噻吩环与烯桥不能很好地共平面,削弱了共轭程度,所以会导致其可见光区吸收峰发生蓝移和缺失精细结构[26]。而同时不论开环体还是闭环体,大位阻体系的摩尔消光系数(ε)均低于小位阻体系的ε。侧端含供电子基团相比于吸电子基团,开闭环体的ε 都相对较高,同时也显示更长的最大吸收波长(表2)。
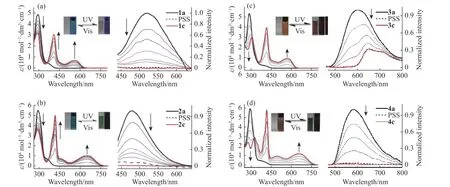
图 6 (a) 1a、(b) 2o、(c) 3a 和(d) 4o 的四氢呋喃溶液在紫外光(λ = (313 ± 10) nm)照射下吸收和荧光光谱的变化。荧光的激发波长分别为(a) 361 nm、(b) 366 nm、(c) 309 nm 和(d) 323 nm。插图展示了颜色和荧光在紫外光(λ = (313 ± 10) nm)和可见光(λ > 470 nm)照射下的变化。图中箭头向上和向下分别表示受到光照后光谱上升或下降的趋势Fig. 6 Absorption and fluorescence changes of (a) 1a, (b) 2o, (c) 3a, and (d) 4o in tetrahydrofuran upon UV irradiation at (313 ± 10) nm.Excitation for fluorescence is set at (a) 361 nm, (b) 366 nm, (c) 309 nm and (d) 323 nm, respectively. Inset images show the color and emission changes triggered by UV (λ = (313 ± 10) nm) and visible (λ > 470 nm) light. The arrows up and down indicate the increase and decrease tendency of the spectra upon irradiation by light, respectively
目标化合物1~4 开环体都具有一定的ICT 效应,表现出一定的荧光性能。当受到紫外光照射(λ =(313 ± 10)nm)时,都发生明显的荧光淬灭(除了3c显示出略微反常的红色荧光),达到光稳态时,荧光淬灭程度与闭环反应转化率基本一致。比较大位阻和小位阻体系可以发现,由于β 位取代甲基增加了噻吩的供电子能力,因此大位阻体系(1a: 519 nm; 3a: 600 nm)的荧光发射波长均长于小位阻体系(2o: 480 nm; 4o:584 nm)。相比较于吸电子基团,供电子基团由于增强了ICT 作用,因此含供电子基团的目标化合物3a 和4o 均表现出波长更长的橘黄色荧光,1a 和2o则呈现出绿色荧光。
在分离得到了目标分子1 和3 的反平行异构体之后,进一步对其进行了量子效率测试。结果发现,1a 和3a 的闭环量子效率均突破了传统二芳基乙烯50% (摩尔分数,下同)的限制,分别为56%和68%(表2),而2o 和4o 的量子效率仅为34% 和32%,符合传统小位阻体系的闭环量子效率。对于开环量子效率,1c 和3c (1c: 11.2%; 3c: 9.0%)均高于2c 和4c(2c: 0.1%; 4c: 1.1%),说明大位阻效应会提高开环量子效率。

表 2 目标化合物1~4 在四氢呋喃溶液中的光谱数据Table 2 Spectroscopic data of compound 1~4 in tetrahydrofuran solution
2.3.2 平行异构体的光致变色性能 在获得了目标分子1 和3 的平行异构体后,对它们的吸收和荧光性能进行测试。从图7 中可以看出,1p 和3p 的吸收与反平行异构体1a 和3a 的吸收基本类似,没有明显区别,除了平行异构体在290 nm 左右的吸收峰和肩峰的强度略强。但是,两者的荧光性能差异反而较大。平行异构体的荧光波长(1p: 482 nm; 3p: 590 nm)要明显短于反平行异构体的波长。而两个平行异构体在受到紫外波长照射后,吸收和荧光均未发生明显的变化,说明平行异构体是一个光惰性的构型。
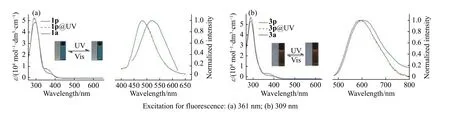
图 7 (a) 1a 和1p (2.09 × 10−5 mol/dm3)和(b) 3a 和3p (2.14 × 10−5 mol/dm3)在四氢呋喃溶液中紫外可见吸收和荧光光谱对比。插图为1p 和3p 在紫外光(λ = (313 ± 10) nm,0.21 mW/cm2,6 min)照射前后的变化Fig. 7 UV-vis absorption and fluorescence spectra of (a) 1a and 1p (2.09 × 10−5 mol/dm3) and (b) 3a and 3p (2.14 × 10−5 mol/dm3) in tetrahydrofuran. Insert images show spectra changes of 1p and 3p upon irradiation by UV light (λ = (313 ± 10) nm, 0.21 mW/cm2) for 6 min
2.4 闭环体热稳定性的研究
目标分子1~4 均具有较优良的光致变色性能,但是其闭环体的热稳定性却各不相同,小位阻的目标化合物2 和4 均能分离得到其各自纯的闭环体化合物2c 和4c,说明其闭环体具有较高的热稳定性。而目标化合物1c 和3c 的热稳定性则较差,两者均无法成功分离,尤其是目标化合物1c。因此进一步研究了目标化合物1c 在不同溶剂中的热稳定性。通过紫外-可见光谱仪中动力学模式测定其在不同溶剂中的衰减曲线,并通过非线性拟合的方式获得其动力学常数(k)和半衰期( τyz)。数据由式 (1) 拟合:

其中:A0、A∞和A 分别代表初始吸光度、最终吸光度和任意时刻的吸光度。目标化合物1c 在不同溶剂中的衰减动力学数据如表3 所示,衰减曲线如图8(a)所示。
通过表3 数据可以发现,目标化合物1c 的热稳定性并不好,在多种溶剂中的热衰减速率均较快,如在甲醇中的半衰期只有371.0 s 左右,而环己烷中其半衰期也仅有2 336 s。说明该化合物的热稳定性较差,同时也可以发现随着溶剂极性的增加,其半衰期也逐渐缩短,热衰减的速率也不断加快,这与文献报道基本一致。1c 较差的热稳定性可以归结于β 位取代甲基与烯桥之间产生较大的空间位阻作用。同时吸电子的吡啶基团使得两个活性碳间碳碳键的电子云偏向吸电子基团,从而削弱了活性碳间单键的键能,进一步地降低了闭环体的热稳定性[29]。同时由于其具有一定的热可逆性,因此它在需要快速热褪色的光致变色眼镜等方面具有潜在的应用[30]。

表 3 293 K 下目标化合物1c 在不同溶剂中的热衰减动力学数据Table 3 Spectrokinetic data of thermal decay for compound 1c in various solvents at 293 K
图8(b)和表4 是目标化合物3c 的热衰减曲线和动力学数据,可以看出目标化合物3c 的热稳定性明显优于目标化合物1c。比较两者的半衰期,可以发现目标化合物3c 的半衰期要比1c 长很多,在四氢呋喃中化合物3c 是1c 的58 倍,而在乙腈中是约90 倍,甲苯中则为40 倍。两者之间的半衰期差值是相当大的,基于类似的核心结构和较大的分子张力,但是闭环体热稳定性却相差这么多,这极有可能与侧端取代基团有关。目标化合物3 中噻吩环所连接的对甲基氧基苯基,具有较好的供电子能力,使得关环反应生成的碳碳键的电子云密度增大,从而进一步地增强了其闭环体的热稳定性[29]。

图 8 293 K 下目标化合物1c(a)和3c(b)在不同溶剂中的衰减曲线Fig. 8 Decay curves of compound 1c(a) and 3c(b) in different solvents at 293 K

表 4 293 K 下目标化合物3c 在不同溶剂中的热衰减动力学数据Table 4 Spectrokinetic data of thermal decay for compound 3c in various solvents at 293 K
通过以上述数据也可看出目标化合物3 的闭环体在不同溶剂中的热稳定性也不同,且也是在极性溶剂中更容易衰减。目标化合物2 和4 则比化合物1 和3 稳定性优异得多。图9 示出了目标化合物1~4 在乙腈中热稳定性的对比图。如图9 所示,在乙腈溶液中,目标化合物2 和4 的闭环体在相当长一段时间内都没有发生明显的热衰减,而目标化合物1和3 则可以看出明显的热衰减趋势,尤其是目标化合物1。热衰减趋势更甚。
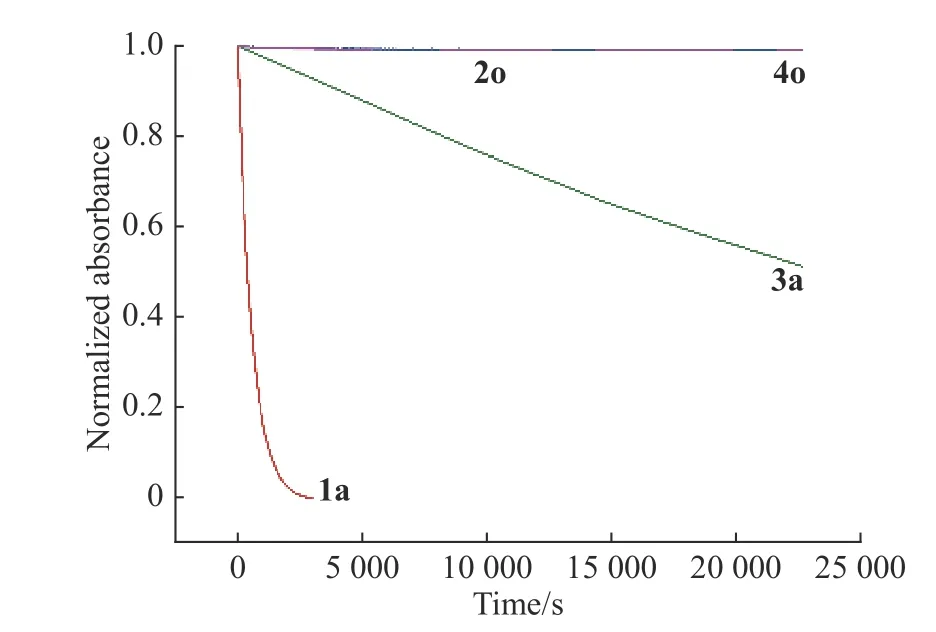
图 9 目标化合物1~4 闭环体在乙腈中的热稳定性对比Fig. 9 Comparison of the thermal stability of closed form of target compounds 1~4 in acetonitrile
热稳定性之所以相差如此之大,一方面是由于目标化合物1c 和3c 的噻吩4 号位的甲基所带来的较大空间位阻效应,另一方面是由于小位阻的2c 和4c 噻吩上氢与烯桥的氮形成氢键作用。
3 结 论
本文基于位阻型烯桥,系统地比较了侧端位阻效应以及噻吩取代基的电子效应对位阻型二芳基乙烯体系光响应性能的影响。通过在噻吩β 位引入甲基取代基,成功分离目标化合物1 和3 的平行与反平行异构体。目标化合物1~4 均具有较好的光致变色性能,其中目标化合物1a 和3a 的闭环量子效率均突破传统二芳基乙烯体系50% (摩尔分数)的限制。但由于所引入甲基造成的空间位阻作用,闭环态1c 和3c 的热稳定性明显降低,同时1c 和3c 两者闭环态的最大吸收波长也较小位阻体系产生了明显的蓝移。并且研究发现1c 和3c 的热衰减速率随着溶剂极性的增加而增加。另外,噻吩取代基的电子效应也会影响热稳定性,含吸电子基团的1c 热衰减速率明显高于含供电子基团的3c。最后,目标化合物1c 和3c 所具有的热可逆性,对于设计和发展快速褪色的光致变色眼镜染料提供了新思路。
猜你喜欢
分子催化(2022年1期)2022-11-02
石油化工(2022年9期)2022-10-19
农业工程学报(2022年12期)2022-09-09
武汉工程大学学报(2022年4期)2022-08-26
西北农林科技大学学报(自然科学版)(2021年1期)2021-03-04
太原理工大学学报(2020年4期)2020-07-22
分析化学(2017年12期)2017-12-25
建筑建材装饰(2016年13期)2017-01-04
山东工业技术(2016年23期)2016-12-23
佛山陶瓷(2016年11期)2016-12-23
