
Ⅱ型Usher综合征或39型视网膜色素变性患者的临床表型、致病基因与新突变位点分析△
2020-12-10 06:16杨极邹悦韦春玲肖丽波焦康为
眼科新进展 2020年12期
杨极 邹悦 韦春玲 肖丽波 焦康为
Usher综合征(USH)是一类常染色体隐性遗传病,常见的临床表型主要为双眼视网膜色素变性、双侧感音神经性听力损害,一般不伴有前庭功能异常。按照疾病临床特点和病程,Usher综合征可分为3型:I型(USH1)、Ⅱ型(USH2)和III型(USH3)。目前已发现16个与Usher综合征存在明确相关性的基因[1],其中74%的Usher综合征为USH2,而30%~40%的USH2患者致病原因为USH2A基因突变。同时,USH2A基因突变也可导致39型视网膜色素变性,其在非综合征性视网膜色素变性患者中占比达10%~15%[2]。USH2A基因突变所致疾病具有显著的遗传异质性[3],目前报道的USH2A基因突变已超过600种[4]。本研究我们采用全外显子测序技术对2个USH2家系和1个39型视网膜色素变性家系进行基因检测,发现3个家系致病原因均为USH2A基因突变,现将结果报告如下。
1 资料与方法
收集 2018年8月至2019年8月就诊于云南省第二人民医院眼科门诊并确诊为视网膜色素变性的先证者3例,其中2例先证者伴有听力损害。详细询问其家族遗传史,绘制家系图,并募集家族中患有同样疾病的近亲属参与研究,对参与研究的成员进行了全面的临床检查,包括:裸眼视力、最佳矫正视力、裂隙灯下眼前段及眼底检查、眼底照相、眼部OCT检查以及荧光素眼底血管造影检查。该研究通过本院医学伦理委员会审核批准,参与研究的患者均知情并签署知情同意书。
抽取参与研究的家族成员静脉血3~5 mL,EDTA抗凝。利用基因组DNA提取试剂盒按标准操作流程提取外周血全基因组DNA。利用Illumina公司定制的基因片段捕获芯片进行靶目标文库富集。文库采用Agilent 2100 Bioanalyzer和ABI StepOne进行片段大小、浓度、富集度的检测。最后利用Illumina Hiseq2000测序仪进行基因测序。将原始测序数据转换为fastq文件后,使用BWA软件将reads比对到人类参考基因组GRCh37/hg19上,生成bam文件[5]。生成的bam文件采用GATK系列软件进行局部重新比对,去除重复序列并进行变异检测[6]。
为锁定可疑致病基因变异,可进行如下分析 :(1)筛选检出变异中的非同义突变进行下一步分析;(2)筛选ExAC EAS、ExAC ALL、1000Genomes、gnomAD等数据库中未见正常人携带或携带率小于5%的变异;(3)参考dbSNP、OMIM、HGMD、ClinVar等多种数据库对致病变异位点进行评估;(4)使用SIFT、PolyPhen2、REVEL等多种蛋白功能预测软件进行基因变异对蛋白功能影响的预测。根据ACMG(2015)分类指南以及患者的临床表型进行致病变异的筛选。对于分析结果为致病或可能致病的位点,采用一代Sanger测序技术进行验证。采用同源序列多重比对,利用UGENE软件分析相关突变位点在不同物种间的保守性[7]。
2 结果
2.1 患者的临床表现根据患者的病史、家族遗传史和临床辅助检测结果,所有的患者眼底检查均表现为视网膜色素变性眼底特点,后极部视网膜可见骨细胞样色素沉积,视网膜血管变细,视盘蜡黄(图1A和图1B)。荧光素眼底血管造影检查可见斑驳状透见荧光、色素斑块所致的荧光遮蔽,视网膜血管一致性变细且有荧光渗漏,周边可见,视网膜毛细血管无灌注所致的充盈缺损(图1C和图1D)。OCT检查可见视网膜变薄,视网膜色素上皮层色素紊乱,感光细胞明显变薄(图1E和图1F)。其中家系1和家系2中患者同时伴有中等程度的听力损害。
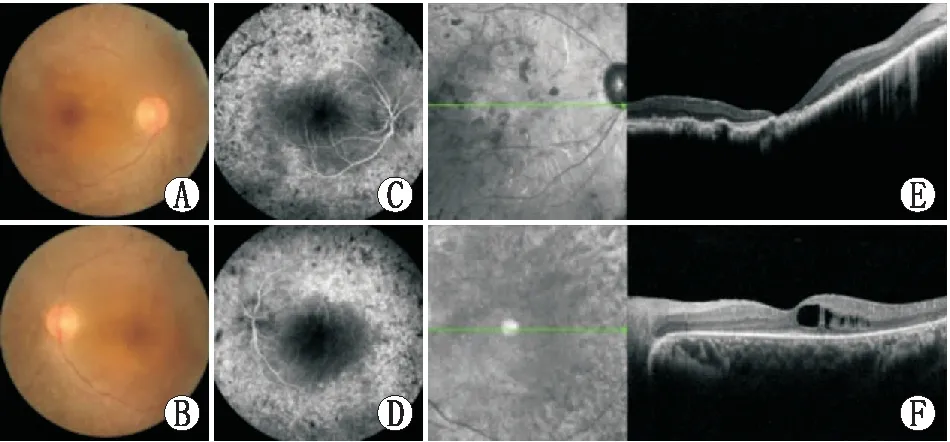
图1 眼底检查结果 A、B:眼底照相显示,后极部视网膜骨细胞样色素沉积,视网膜血管变细,视盘蜡黄;C、D:荧光素眼底血管造影显示斑驳状透见荧光,可见色素斑块沉积所致的荧光遮蔽,视网膜血管一致性变细且有荧光渗漏,周边可见,视网膜毛细血管无灌注所致的充盈缺损;E、F:OCT检查可见视网膜变薄,视网膜色素上皮层色素紊乱,感光细胞变薄
2.2 患者的基因特点家系1先证者F1-II1(图2), 男性,诉双眼视力下降伴夜盲、耳聋12 a。家系1先证者患病的弟弟F1-II2,男性,诉双眼夜视力下降1 a。2例患者眼底检查均出现典型的视网膜色素变性眼底改变,结合病史和辅助检测,高度怀疑患有USH2。基因检测结果显示,F1-II1和F1-II2的USH2A基因42号外显子上均存在c.8232G>C (p.W2744C)(M1)纯合错义突变(图3A)。M1突变Polyphen2的HumDiv分值为1.0,HumVar分值为1.0。保守性分析显示,c.8232G>C (p.W2744C)所对应氨基酸位点在多物种间高度保守。3D蛋白预测模型提示,c.8232G>C (p.W2744C)突变影响蛋白质的局部构像。HGMD数据库信息收录显示,M1突变曾在常染色体隐性遗传的 USH2患者中被报道,且该突变在正常人群数据库中未见收录。家系共分离验证结果显示,F1-I1和F1-I2均携带M1的杂合突变,F1-II1和F1-II2的M1纯合突变分别来自其健康的父母(图3B)。依据ACMG指南的致病性分析,M1突变满足致病证据PS1+PM2+PP,可判断该突变为致病性变异。结合基因检测结果,最终确诊为USH2。
家系2先证者F2-II1(男性)和其患病的妹妹F2-II2 (图2),均有双眼视力下降伴夜盲、耳聋的病史。基因检测结果显示,F2-II1和F2-II2均于USH2A基因的42号内含子内存在c.8559-2A>G(M2)纯合剪接突变(图3C)。M2突变为剪接位点突变,HSF剪接突变分析显示,Site broken变异系数为-34.4%,提示该突变可导致受体位点的改变,影响剪接。保守性分析显示,43号外显子编码的氨基酸位点2853在多物种间高度保守。HGMD数据库信息收录显示,M2突变曾在USH2患者中被报道,该突变在正常人群数据库中的携带频率为0.000 3,属于低频突变。家系共分离验证结果显示,F2-I1和F2-I2均携带M2杂合突变,即F2-II1和F2-II2的M2纯合突变分别来自其健康的父母。依据ACMG指南,M2突变满足证据PVS+PS1+PM2,可判断该突变为致病性变异。结合基因检测数据,最终确诊为USH2。
家系3先证者F3-II1 (图2),男性,诉双眼夜视力下降,辅助检查和临床表现提示视网膜色素变性,患者无听力受损。基因检测在USH2A基因上发现c.12778C>T(p.Q4260X)(M3)纯合无义突变(图3D)。无义突变可导致蛋白质合成中断,进而影响基因功能。该突变在正常人群数据库中未见收录。HGMD数据库未收录M3突变。依据ACMG指南,M3满足证据PVS1+ PM2,可判断该突变为疑似致病性突变。由于患者父母已故,未能进行家系共分离验证。结合基因检测,最终确诊为39型视网膜色素变性。

图2 患者家系图 箭头所指为先证者
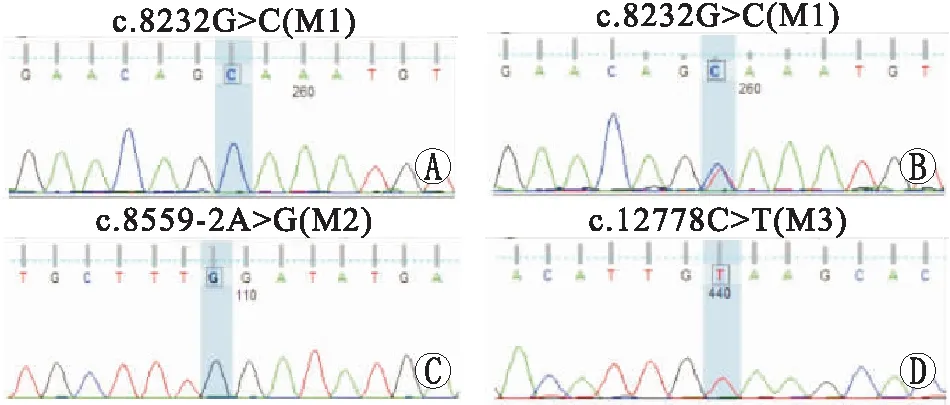
图3 Sanger测序图 A:家系1先证者F1-II1和患者F1-II2检测出USH2A基因上存在c.8232G>C (p.W2744C)(M1)的纯合错义突变; B:家系1中 F1-I1、F1-I2检测出USH2A基因上存在c.8232G>C (p.W2744C)(M1)的杂和错义突变;C:家系2中先证者 F2-II1和患者F2-II2检测出c.8559-2A>G(M2)纯合剪接突变;D:家系3中先证者F3-II1检出c.12778C>T(p.Q4260X)(M3)纯合无义突变
3 讨论
本研究中3个家系均具有不同的病程及临床症状,家系1和家系2的所有患者眼底检查呈典型的视网膜色素变性改变,同时伴有中度感音神经性耳聋。家系3中的患者表现为单纯视网膜色素变性,并无耳聋的临床表现。基因检测结果提示,5例患者的致病基因均为USH2A基因,此基因突变可导致Ⅱ型USH或39型视网膜色素变性,结合患者临床症状、病程和基因检测结果,家系1和家系2的所有患者均诊断为USH2,家系3中患者诊断为39型视网膜色素变性。USH2A基因位于人类常染色体1q41,由72个外显子组成,编码USH2A蛋白,此蛋白包括异构体A和异构体B。USH2A蛋白异构体A被认为是外分泌蛋白[8]。异构体B主要分布于视网膜,除了异构体A的所有组分,还包括2个LamGL区域、 28个FN3区域、 1个跨膜序列以及1个胞内区,此胞内区C端有一个PDZ结合区域[9]。USH2A蛋白位于光感受器细胞的外周纤毛区,组成USH2蛋白复合体,为外周纤毛和连接纤毛提供结构支持,参与感光细胞的发育和维持感光细胞内节到外节的物质运输[10]。目前在USH2A基因上发现的各类突变已超过600个,包括:错义突变、无义突变、剪接突变和移码突变[11]。USH2A基因所致疾病具有高度遗传异质性和临床异质性,即使为同一位点突变,患者的临床表现也不一定相同,部分患者表现为视网膜色素变性,也有部分患者表现为USH2,故对视网膜色素变性和USH2患者进行基因检测明确病因具有重要意义[12]。
本研究中,我们应用全外显子测序技术对5例患者及其父母进行了基因检测。通过基因检测发现,在USH2A基因上存在c.8232G>C (p.W2744C)(M1)、c.8559-2A>G(M2)和c.12778C>T(p.Q4260X)(M3)突变,其中M3突变为首次报道。M1突变在dbSNP、ExAC Browser、the 1000 Genomes Project等数据库中为低频变异。蛋白同源性序列分析发现突变位点在多物种中均具有高度保守性,同时Mutation Taster、SIFT和Polyphen2预测软件分析证实具有致病性。三维蛋白结构预测模型提示,突变影响了蛋白质的稳定性,可影响蛋白跨膜段,影响USH2A蛋白与膜的整合,影响感光细胞的物质转运。M2为纯合剪接突变,保守性分析显示,对应氨基酸片段在多物种间高度保守,剪接突变可导致基因外显子翻译功能受到影响,进而导致USH2A蛋白功能发挥障碍。M3为纯合无义突变,该突变使蛋白编码提前终止,进而影响其功能。
由于USH2A基因突变所致疾病具有的高度遗传异质性,目前对其基因型和表现型间关联的了解仍然较少,对于USH2A突变体蛋白在视网膜中所发挥的作用了解依然有限。在所有已知的USH2致病基因中,USH2A基因是最为常见的致病基因,它所编码蛋白质对感光细胞的发育和功能发挥具有不可替代的作用[13],同时USH2A基因突变也可导致39型视网膜色素变性。本研究中的三个家系均由USH2A基因突变致病,但临床表型存在差异。不同的变异位点所致的蛋白合成和翻译变异不同,对蛋白功能影响作用也不尽相同。因此,对USH2A基因突变的鉴定不仅可以阐明其在USH2和39型视网膜色素变性发病中的作用,而且有助于临床诊断和寻找治疗疾病的有效方法[14]。本研究结果扩大了对USH2A基因突变谱的认知,进一步阐明其致病的分子机制,有利于提高该疾病诊断的准确率[15]。
猜你喜欢
中国造纸(2022年9期)2022-11-25
中国造纸(2022年8期)2022-11-24
中华实用诊断与治疗杂志(2022年1期)2022-08-31
临床输血与检验(2022年3期)2022-06-22
诊断学(理论与实践)(2020年1期)2020-04-28
郑州大学学报(医学版)(2019年3期)2019-06-03
传染病信息(2019年2期)2019-05-17
环球时报(2019-04-03)2019-04-03
吉林林业科技(2018年6期)2018-11-21
听力学及言语疾病杂志(2015年5期)2015-12-24
