
先天性胆汁酸合成障碍2型8例临床特征及基因变异分析
2020-12-23 06:54佘兰辉李旭芳叶家卫谭丽梅杨花梅房春晓
临床儿科杂志 2020年12期
佘兰辉 李旭芳 叶家卫 谭丽梅 杨花梅 房春晓 龚 余 徐 翼
广东省广州市妇女儿童医疗中心(广东广州 510120)
胆固醇合成胆汁酸的途径涉及16个酶催化的17步反应,其中任何一步障碍均可导致胆汁酸合成障碍,中间产物在肝脏及肝外组织堆积,在不同年龄阶段出现不同的疾病谱。其中δ-4-3-氧固醇-5β-还原酶(δ 4-3-oxosteroid 5 β-reductase deficiency,AKR 1 D 1)将胆固醇核转变成胆汁酸核,其缺陷引起的临床表型称为先天性胆汁酸合成障碍2型(congenital bile acid synthesis defect type 2,CBAS2),后者可引起严重的肝内胆汁淤积症,于婴儿期甚至新生儿期起病,表现为持续黄疸不退、严重胆汁淤积、排浅色或白陶土样粪便,可有脂肪泻,伴肝脾肿大及生长发育迟缓,可因维生素K1吸收不良出现凝血功能障碍,部分早期即进展为肝衰竭。1988年首次在同卵双胎男孩中发现CBAS 2[1],此后相续出现多个病例[2-6]。生化检查显示丙氨酸氨基转移酶及天冬氨酸氨基转移酶明显升高,伴有高胆红素血症,以结合胆红素升高为主,不伴瘙痒,而血清γ-谷氨酰转肽酶(γ-glutamyl transpeptidase,γ-GT)和总胆汁酸正常或偏低。尿液类固醇分析发现大量7 α-羟3-氧-4-胆烷酸和7 α,12 α-二羟-3-氧-4-胆烷酸。这些患儿可在婴儿期甚至新生儿期因暴发性肝衰竭或多器官功能障碍综合征而死亡。CBAS2较罕见,早期可因浅黄色甚至白陶土样大便误诊为胆道闭锁,亦有部分可因凝血障碍、血氨基酸及尿代谢产物异常而被误诊为Citrin缺陷导致的新生儿肝内胆汁淤积症(neonatal intrahepatic cholestasis caused by Citrin deficiency,NICCD)。早期通过尿类固醇及血基因检测可明确CBAS2诊断,而早期 干预可改善预后,故有必要总结其临床特征、生化特征。
1 临床资料
回顾分析2018年1月至2020年3月广州市妇女儿童医疗中心诊断的CBAS 2患儿的临床资料。共纳入8例患儿,分别来自8个无血缘关系的家族,其中6例男性、2例女性,均为广东籍。父母均为非近亲结婚,除例2患儿为G4P4,第1胎肝衰竭死亡(具体不详),均否认家族史。8例患儿出生均为足月,出生体质量3.0~3.6 kg,平均体质量(3.2±0.2)kg,均以黄疸持续不退就诊,初次就诊年龄为1.5~3个月,中位时间为2个月。初次肝功能检测年龄均为满月后,在1~3个月之间,中位时间为49天。诊断CBAS2年龄为3~7个月,中位诊断年龄为4.7个月,初次就诊时体质量增长均可。见表1。
8例患儿均以黄疸为主要临床表现,出现黄疸的中位年龄为3.0(2.0~3.0)天。4例患儿于新生儿期接受蓝光治疗,黄疸稍有减轻但未完全消退。所有患儿满月后仍持续黄疸,并进行性加深,大便颜色偏浅。6例大便浅黄色,其中3例泛白色甚至呈白陶土样大便。体格检查:生长发育无异常,无异常面容。7例患儿的肝右肋下达3 cm及以上,有5例伴脾大。胆囊彩色超声均提示空腹胆囊充盈差或不充盈。其中7例患儿行磁共振胰胆管成像(magnetic resonance cholangiopancreatography,MRCP)检查,6例提示肝内胆管细小,左右肝管汇合远端、肝总管及胆总管可见;另1例患儿因疑似胆道闭锁已行腹腔镜胆道探查及肝活检,病理提示肝细胞气球样变,胞浆内淤胆明显,汇管区结构存在,未见小胆管增生,未见纤维增生,少量淋巴细胞浸润。
8例患儿在初级胆汁酸干预前,生化指标均提示丙氨酸氨基转移酶、天冬氨酸氨基转移酶、碱性磷酸酶、甲胎蛋白升高,胆汁酸水平不高或升高不明显(<13 μmol/L),γ-GT不高或轻度升高(2例轻度升高,60~120 U/L)。8例患儿的胆红素均明显升高,以直接胆红素升高为主,白蛋白及血氨水平均无异常。2例患儿的凝血酶原时间(prothrombin time,PT)稍延长16~17 s,1例延长>20 s,余5例凝血功能大致正常。6例患儿行血氨基酸及血酰基肉碱检测,其中5例血氨基酸分析均提示苏氨基、酪氨基、蛋氨基等多种氨基酸含量明显升高;4例血酰基肉碱无异常,2例酰基肉碱检测提示多种长链酰基肉碱含量偏高。8例患儿行尿气-相色谱(gas chromatography-mass spectrometer,GC/MS)检测,2例尿中见中量至大量的中量4-羟基苯丙酮酸,4-羟基苯乳酸。
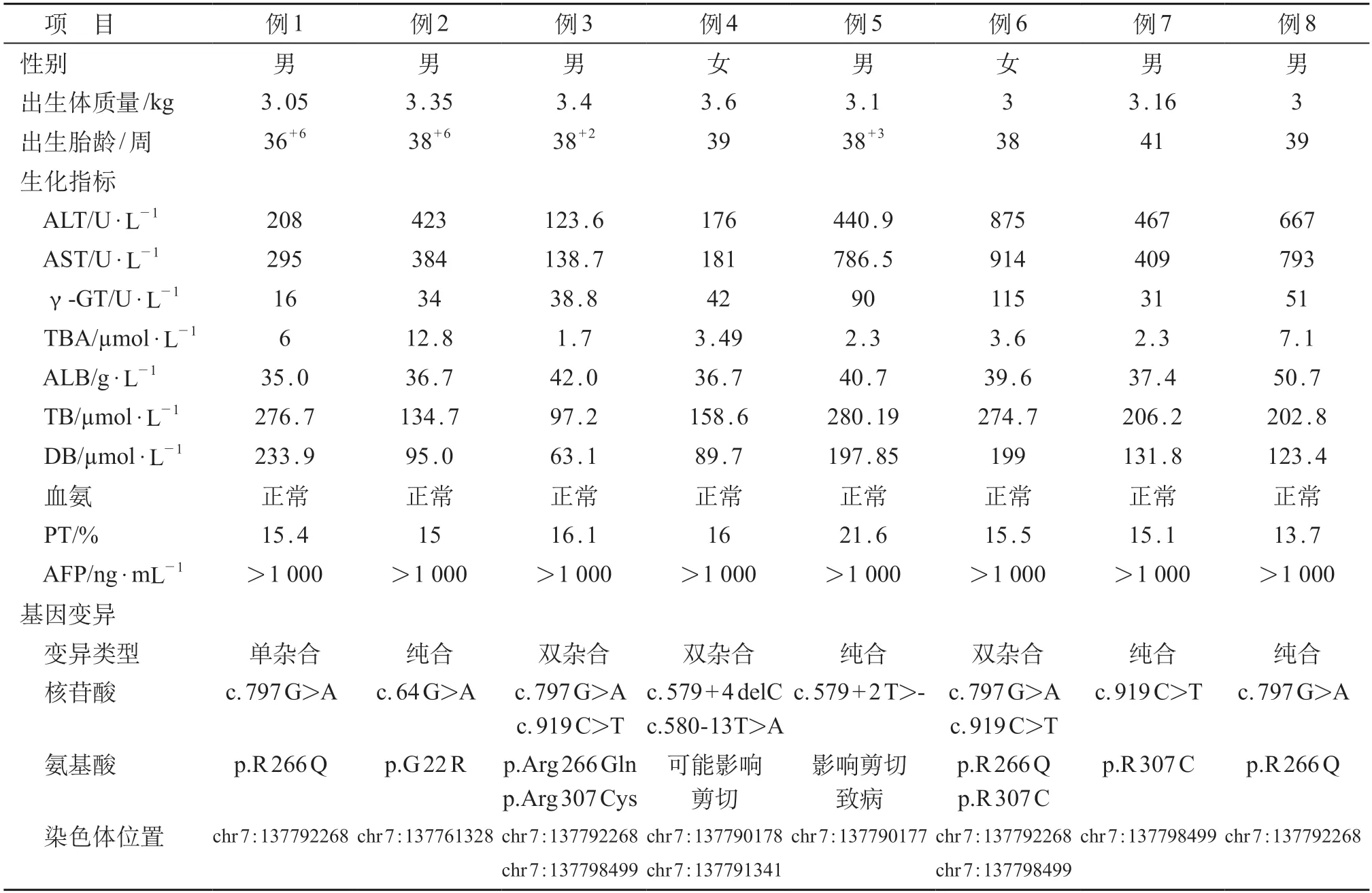
表1 8例患儿一般情况、生化及AKR1D1基因变异情况
8例患儿均符合胆汁淤积性肝病诊断:具有黄疸,伴或不伴肝脏肿大或质地改变,伴有丙氨酸氨基转移酶(alanine aminotransferase,ALT)升高,总胆红素(total bilirubin,TBil )>85 μmol/L,且直接胆红素(direct bilirubin,DBil)/Tbil>20%。8例患儿经常规护肝、退黄、利胆治疗,效果欠佳,影像学/腹腔镜胆道探查排除胆道闭锁,结合胆汁酸及γ-GT不高,考虑遗传代谢性疾病特别是胆汁酸合成障碍可能。经医学伦理委员会审核,家长知情同意后,抽取患儿及父母2~3 mL EDTA抗凝血,送至康圣达基因公司行黄疸相关遗传医学外显子检测。首先采用高通量测序的方法检测基因的变异情况,再按照2015年美国医学遗传学和基因组学学院(American College of Medical Genetics and Genomics,ACMG)的基因变异解读标准和指南做致病性分析,最后运用Sanger测序技术对疑似致病变异进行验证。结果提示:8例患儿均有AKR 1 D 1基因变异;其中4例是纯合变异,3例复合杂合变异,1例单杂合变异;单杂合变异患儿结合尿类固醇分析亦诊断明确。8例患儿的变异位点包括移码变异(c.579+4 delC,c.579+2T>-)和错义变异(c.797 G>A,c.64 G>A,c.919 C>T,c.580-13 T>A,c.3362 A>G)。其中2例基因型一致(复合杂合c.797G>A和c.919C>T)。
2 讨论
CBAS2是一种常染色体隐性遗传病,由AKR1D1缺陷引起,后者属于醛酮还原酶家族1成员D 1 。编码AKR1D1基因位于人类7号染色体q32-q33区[3],全长42 kb,共包含9个外显子,主要表达于肝脏。AKR1D1以烟酰胺腺嘌呤二核苷酸磷酸为氢化物供体,可催化具有3-氧代-4-烯结构的类固醇还原,减少胆汁酸中间体a环中的双键,最终产生初级胆汁酸鹅去氧胆酸(chenodeoxycholic acid,CDCA)和胆酸(cholic acid,CA)[4],因而在胆固醇合成初级胆汁酸过程中非常重要。AKR1D1缺陷导致3-氧代-4-烯结构的类固醇聚集,CDCA及CA合成受碍,影响正常胆汁流,肝功能受损,异常中间代谢产物堆积,尿异常有机酸堆积,故可出现病例中所表现的黄疸,浅色便,肝肿大,如未干预后续可出现营养不良,发育迟缓,渐进展至肝硬化、肝衰竭。AKR1D1的缺乏除先天性基因缺陷所致的CBAS2外还包括继发性5β还原酶缺陷,后者包括引起暴发性肝衰竭的各种病因,包括新生儿血色素沉着症、酪氨基血症1型[3,5]。这些疾病均表现为γ-GT和胆汁酸正常,同时尿中3-氧-δ4酸含量增加,因而不能通过γ-GT和胆汁酸来区分开先天性及继发性。有研究表明,可通过检测尿类固醇水平将血色病及先天性胆汁酸缺乏与其他继发性5b-还原酶缺乏症相鉴别[6],而基因分析是唯一可靠的方法。自1997年开始行基因检测[7],目前已报道的基因确诊病例有30余例[2-7]。本组8例患儿的变异类型中,c.797G>A、c.919C>T为已报道的致病变异[8,10]。c.579+4 delC和c.580-13 T>A复合杂合变异,HGMDpro数据库暂无报道,参考 ACMG基因变异解读指南[9],可能影响剪切-pp3而致病。c.64G>A及c.579+2T>-,HGMD专业版数据库中无收录,参考ACMG基因变异解读指南分级分别为可能致病及致病,结合临床表现及生化检测结果明确诊断。本组8例患儿中,c.797 G>A和c.919C>T有重复出现,携带率分别为5/16、4/16,均高于上海研究[10]。本组患儿中1例为AKR1D1基因单杂合变异,结合影像排除继发性因素,进一步完善快速原子轰击电离质谱对尿类固醇进行检测分析,最终明确CBAS2诊断。故在CBAS2的诊断中,尿液类固醇分析和基因检测都非常重要。
CBAS2的初级胆汁酸合成障碍,CDCA和CA合成减少,而甘氨酸和葡萄糖醛酸结合的胆汁酸缺乏,致胆囊充盈欠佳及胆囊小,且因肠道内粪胆原减少,大便颜色浅甚至为白陶土样大便,因而易误诊为先天性胆道闭锁,但后者一般胆汁酸及γ-GT明显升高,因而当出现浅色大便,且胆囊充盈差、小胆囊时,需注意胆汁酸及γ-GT水平,当后两者均不高或升高不明显时需警惕先天性胆汁酸合成障碍的可能,尽早行尿类固醇和基因检测,避免胆道探查等有创性检查。在正常胆汁流受影响后,异常中间产物在肝内堆积,影响肝脏功能,蛋白及脂代谢受影响,可出现血瓜氨基、酪氨基、蛋氨基等多种氨基酸含量明显升高,尿中可见中量至大量的4-羟基苯丙酮酸,4-羟基苯乳酸,易误诊为NICCD,如本组患儿中5例血氨基酸改变及2例尿GC/MS改变,但因NICCD可伴有颜面丰满、低蛋白、高乳酸,且转氨酶升高以谷草转氨酸升高为主,结合基因可鉴别。
目前CBAS 2治疗主要是初级胆汁酸替代治疗,通过替代治疗,补充人体必需的初级胆汁酸,对异常胆汁酸合成起负反馈作用,减少毒性中间代谢产物在缺陷肝细胞异常的产生。目前研究表明CA[11]、CDCA[7]及熊去氧胆酸均可缓解症状及延缓肝脏损害。但熊去氧胆酸虽可改善生化、影像学及组织学指标,但不能降低内源性胆汁酸合成,不能防止不典型或潜在肝毒性胆汁酸中间体及其代谢产物的合成,不能降低尿有机酸的水平,因而不推荐使用。目前推荐治疗为CA及CDCA,研究报道替代治疗后肝功能好转、黄疸明显消退[7]。
综上所述,婴儿期甚至新生儿期出现黄疸,大便浅黄色,胆红素和转氨酶升高但胆汁酸及γ-GT不高或升高不明显时,需警惕CBAS2的可能,避免误诊为胆道闭锁或NICCD或其他引起胆汁淤积的病因,可通过尿液类固醇和基因检测确诊,尽早初级胆汁酸替代治疗。
猜你喜欢
现代临床医学(2022年4期)2022-09-29
种子(2021年3期)2021-04-12
中西医结合肝病杂志(2020年2期)2020-10-27
心肺血管病杂志(2018年11期)2018-12-18
中国生育健康杂志(2018年6期)2018-11-13
分析化学(2017年12期)2017-12-25
科技视界(2016年27期)2017-03-14
中学生理科应试(2016年7期)2016-05-14
中华老年多器官疾病杂志(2016年8期)2016-05-14
中国卫生标准管理(2015年16期)2016-01-20
