
高效液相色谱荧光检测法测定药物中的氯乙酰氯
2021-01-25 07:47汤加园陈向明
化学分析计量 2021年1期
汤加园,陈向明
(滨州医学院药学院,山东烟台 264003)
药物中的遗传毒性杂质是一类能损伤细胞遗传物质并引起基因突变或体内诱变的物质,较少的摄入量就能够对身体健康产生严重的损害[1-2]。近年来,在市场销售的某些药物中发现了遗传毒性杂质残留,对患者的健康造成威胁,并使相关医药企业遭受巨大经济损失,因此需要对其在药物中的残留量进行严格控制[3-4]。欧洲EMA、美国FDA 和ICH相继发布了遗传毒性杂质限量的指南,建议用毒理学关注阈值(Threshold of toxicological concern,TTC)作为遗传毒性杂质的可接受限度,即日摄入量不超过1.5 μg 是可接受的(其致癌风险小于十万分之一)[5]。中国药典2020 年版中,新增了《遗传毒性杂质控制指导原则》[6],确定了遗传毒性杂质危害评估方法、可接受摄入量计算方法和限度制定方法,为药物中遗传毒性杂质的控制提供了技术指导。
脂肪酰氯类化合物是许多药物合成的重要原料,其分子中的酰氯官能团是遗传毒性杂质的警示结构,具有潜在的遗传毒性,需要严格控制其在药物中的残留量[7]。脂肪酰氯的测定方法包括气相色谱法[8-9]和高效液相色谱法[10-11]。由于脂肪酰氯的高反应活性,无论采用哪种色谱方法,都需要在进样前进行衍生化处理。常用的衍生试剂包括水[12-13]、甲醇[13]、哌啶[14]等。但是这些方法存在灵敏度低、准确度差等缺点,从而限制了方法的应用。高效液相色谱-荧光检测法也是测定样品中痕量成分的常用分析技术[15],其灵敏度和选择性显著优于高效液相色谱-紫外检测法[16]。
笔者以吖啶酮乙酰肼(AHAD)为柱前荧光衍生试剂,建立了测定药物中氯乙酰氯含量的方法。通过衍生化降低了氯乙酰氯的活性,提高了检测灵敏度。所建立的方法成功应用于氟伐他汀钠原料药中氯乙酰氯的含量测定。
1 实验部分
1.1 主要仪器与试剂
高效液相色谱仪:Agilent1260 型,配G1311B型四元梯度泵,G1329B 型自动进样器,G1316A 型恒柱温箱,G1321B 型荧光检测器,美国安捷伦科技有限公司。
氯乙酰氯对照品:纯度为98%,上海阿拉丁生化科技股份有限公司。
氟伐他汀钠原料药:纯度为98%,嘉兴思诚化工有限公司。
吖啶酮乙酰肼:本实验室自制。
乙腈:色谱纯,天津市福晨化学试剂厂。
实验用水为娃哈哈纯净水。
实验用其余试剂均为分析纯。
1.2 溶液的配制
氯乙酰氯标准溶液:精确量取205 μL 氯乙酰氯对照品,置于25 mL 容量瓶中,用无水乙腈溶解并稀释到标线,摇匀,作为氯乙酰氯的标准储备液,浓度为0.1 mol/L。低浓度氯乙酰氯工作溶液用无水乙腈稀释而得。
衍生试剂溶液:精密称取1.34 mg 吖啶酮乙酰肼,用无水乙腈溶解后,定容至100 mL,浓度为5×10-5mol/L。
所有溶液均密封保存于4℃冰箱中。
1.3 衍生过程
分别将100 μL 氯乙酰氯标准溶液或样品溶液和120 μL 吖啶酮乙酰肼溶液,依次加入到1.5 mL离心管中,在室温下衍生15 min。衍生反应概况如图1。反应结束后,向离心管中加入780 μL 50%乙腈溶液稀释后进样。

图1 衍生反应概况
1.4 色谱条件
色谱柱:Agilent ZORBAX Eclipse XDB-C18(250 mm×4.6 mm,5 μm);柱温:30 ℃;流动相:A 为水,B 为乙腈;梯度洗脱程序:0~20 min 时,B 由20%升至40%,流量为1.0 mL/min;荧光激发波长和发射波长分别为255 nm 和429 nm;进样体积:10 μL。
2 结果与讨论
2.1 色谱条件的优化
为实现良好的色谱分离和较短的分析时间,对色谱条件进行了优化。本研究比较了Agilent ZORBAX SB-C18(150 mm×4.6 mm,5 μm) 和Agilent ZORBAX Eclipse XDB-C18(250 mm×4.6 mm,5 μm)色谱柱,结果表明,衍生产物在XDB-C18色谱柱峰形更好,与衍生试剂和副产物的峰均能基线分离,以乙腈和水为流动相,采用梯度洗脱,分析时间能控制在15 min 内。因此选择Agilent ZORBAX Eclipse XDB-C18色谱柱对衍生产物进行分离。
2.2 衍生条件的优化
氯乙酰氯本身无荧光,且酰氯基团具有高反应活性。通过衍生化引入荧光基团以提高检测灵敏度,同时能抑制氯乙酰氯的高活性。酰氯类化合物能与氨基快速的发生反应,因此本研究选择实验室自制的吖啶酮乙酰肼为柱前衍生试剂。
氯乙酰氯与吖啶酮乙酰肼的衍生反应无需催化剂即能发生。为达到最高的衍生效率,实验研究了温度、时间和衍生试剂的用量对衍生反应的影响,结果见图2~图4。由图2 可知,衍生反应在室温下能快速发生,提高反应温度,衍生产物的色谱峰面积略有下降,可能是因为较高的温度导致副反应的发生,因此实验选择室温下进行衍生化。由图3 可知,在15 min 时色谱峰面积达到最大,延长反应时间,色谱峰面积没有明显变化,因此选择15 min 的衍生时间。为保证衍生反应进行的完全,衍生试剂需要过量,由图4 可知,当衍生试剂的浓度是氯乙酰氯浓度的5 倍时,即衍生试剂体积为100 μL 时,色谱峰面积达到最大,继续增加衍生试剂的用量,衍生产物的色谱峰面积没有明显变化,因此衍生反应时,衍生试剂的用量至少是氯乙酰氯浓度的5 倍。

图2 温度对衍生反应的影响
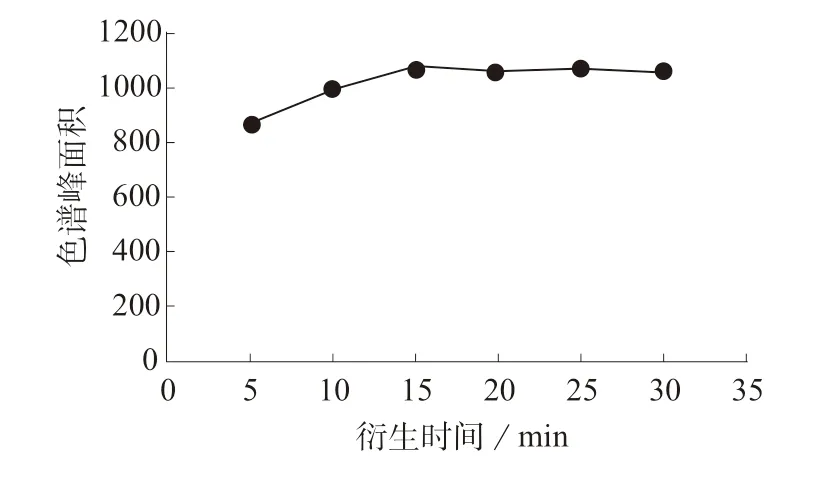
图3 衍生时间对衍生反应的影响

图4 衍生试剂体积对衍生反应的影响
2.3 线性关系和检出限
取氯乙酰氯标准储备液用无水乙腈稀释得到一系列不同浓度的标准溶液,按照前述方法衍生后进样分析,记录色谱图(见图5)和色谱峰面积。
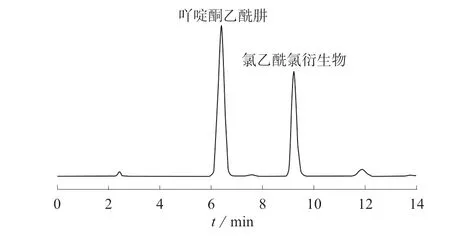
图5 氯乙酰氯标准溶液衍生色谱图
以峰面积作为纵坐标(Y),浓度作为横坐标(X),进行线性回归,氯乙酰氯的浓度在1~1 000 nmol/L范围内与色谱峰面积呈现良好的线性,线性方程为Y=0.992 1X-0.770 6,相关系数r=0.999 9。以信噪比S/N=3 求得方法检出限为0.35 nmol/L。
2.4 精密度试验
将浓度为1×10-5mol/L 氯乙酰氯标准液,按照前述方法衍生后重复进样6 次测定,分别记录峰面积,计算相对标准偏差,如表1 所示,峰面积测定结果相对标准偏差为0.52%,说明仪器性能良好。取6 份浓度为1×10-5mol/L 氯乙酰氯标准液,按照前述方法分别衍生进样测定,结果见表1,峰面积测定结果相对标准偏差为0.67%,说明该方法精密度良好。

表1 仪器精密度和方法精密度试验
2.5 稳定性试验
将标准氯乙酰氯衍生溶液于室温下保存,分别在0、2、4、8、12、24 h 时进样测定,记录峰面积,进行稳定试验,结果如表2 所示,计算得RSD 为3.48%,说明衍生产物在室温保存下24 h 内稳定。
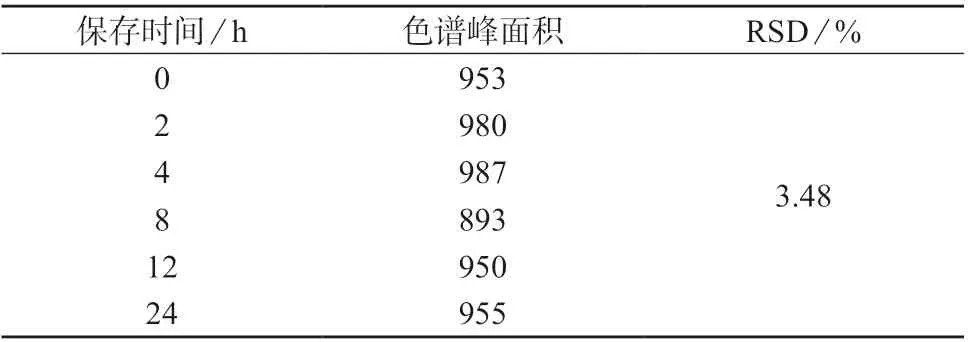
表2 氯乙酰氯衍生物的稳定性试验
2.6 样品的测定
精密称取20 mg 氟伐他汀钠原料药于1.5 mL离心管中,加入1 mL 无水乙腈,超声处理10 min后离心。取上清液100 μL,按照前述方法衍生进样,未发现所测氟伐他汀钠原料药中残留氯乙酰氯。
2.7 加标回收试验
精密称取20 mg 氟伐他汀钠原料药于1.5 mL离心管中,加入高、中、低三种浓度氯乙酰氯标准溶液,每种浓度制备三份样品。按照前述样品处理方法提取衍生后进样分析,记录色谱图,结果见表3。

表3 加标回收试验结果
由表3 可知,样品加标回收率为92.5%~95.6%,说明该方法具有较高的准确度。样品加标色谱图见图6。

图6 氟伐他汀钠原料药加氯乙酰氯标准溶液衍生色谱图
3 结语
采用吖啶酮乙酰肼为衍生试剂对氯乙酰氯进行柱前衍生化,建立了测定氟伐他汀钠原料药中氯乙酰氯的高效液相色谱方法。研究探索了温度、时间和衍生试剂的用量对衍生反应的影响,发现衍生试剂过量5 倍时在室温下反应15 min,衍生产率最大。该方法具有灵敏度高、选择性好、分析速度快的优点,能准确测定氟伐他汀钠原料药中氯乙酰氯的含量,可用于氟伐他汀钠原料药的质量控制。
猜你喜欢
煤化工(2022年3期)2022-07-08
中国经济周刊(2021年22期)2021-12-07
合成纤维工业(2021年5期)2021-10-31
现代仪器与医疗(2021年1期)2021-06-09
保健文汇(2020年8期)2020-12-02
首都食品与医药(2020年1期)2020-10-21
环球时报(2020-02-20)2020-02-20
传媒评论(2019年6期)2019-10-14
中国经济信息(2017年17期)2017-09-09
山东工业技术(2016年10期)2016-09-06
