
液相色谱—串联质谱法测定大米、小麦粉中灭草松及其代谢物残留量
2021-01-25 05:23余婷婷王会霞
食品与机械 2021年1期
刘 迪 杨 青 韩 莉 余婷婷 曹 琦 江 丰 王会霞
(1.湖北省食品质量安全监督检验研究院,湖北 武汉 430075;2.湖北省食品质量安全检测工程技术研究中心,湖北 武汉 430075)
灭草松(Bentazone)又名排草丹、苯达松[结构式见图1(a)],为巴斯夫公司开发的苯并噻二嗪酮类除草剂,是一类兼触杀性和内吸性除草剂[1-3],被广泛应用于芽后杂草的防除,可用于水稻、小麦田中除草,对绝大多数双子叶植物和莎草科杂草有杀除作用,对禾本科植物无害[4]。灭草松属于低毒或中等毒性农药,其在水稻和小麦等植物中会被还原代谢成6-羟基灭草松[图1(b)]或8-羟基灭草松[图1(c)],而相应的代谢产物6-羟基灭草松和8-羟基灭草松均具有毒性[8],对人体有一定的毒性,一般水体污染比较严重[5-7]。
根据GB 2763—2019规定,灭草松残留物定义为灭草松、6-羟基灭草松及8-羟基灭草松之和,以灭草松表示,稻谷和麦类的临时最大残留限量为0.1 mg/kg,且没有规定法定检测方法。目前,关于灭草松的测定方法有气相色谱法和气质联用法[9-10]、离子色谱法[11-12]、液相色谱法[13-14]和液质联用法[15-18]。实际检测过程中,气相色谱及气质联用法一般需要衍生化处理,过程比较繁复;离子色谱法应用相对较少,同时色谱柱的选择性相对较弱;液相色谱法则存在样品基体干扰大;而液相色谱串联质谱法灵敏度高,选择性强。同时现有的灭草松残留检测建立方法多见灭草松母体,而代谢物6-羟基灭草松和8-羟基灭草松检测方法只有液相方法的报道[4],该方法灵敏度和准确度不高。目前能同时测定灭草松及其代谢物6-羟基灭草松和8-羟基灭草松的液相色谱串联质谱方法还未见报道。试验拟以大米和面粉为研究对象,对QuEChERS法提取和净化方法、液相质谱参数等作优化处理,选择C18吸附剂对样品作分散固相萃取净化,建立大米和面粉中灭草松及其代谢物6-羟基灭草松与8-羟基灭草松的液相色谱—串联质谱检测方法。
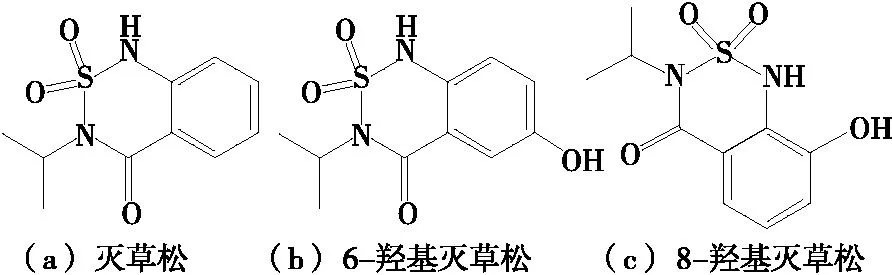
图1 灭草松及其代谢物结构式Figure 1 Structure formula of Bentazone and its metabolites
1 试验部分
1.1 材料及试剂
灭草松(CAS号:25057-89-0)、6-羟基灭草松(CAS号:60374-42-7)、8-羟基灭草松(CAS号:60374-43-8)标准品:纯度>98%,德国DR公司;
大米、小麦粉:市售;
乙腈(CAS号:75-05-8)、甲醇(CAS号:67-56-1):色谱纯,德国默克公司;
乙酸(CAS号:64-19-7)、甲酸(CAS号:64-18-6):质谱纯,美国Fisher公司;
无水硫酸钠(CAS号:15124-09-1)、无水硫酸镁(CAS号:7487-88-9):优级纯,国药集团化学试剂有限公司;
乙二胺-N-丙基硅烷化硅胶(PSA):40~60 μm,博纳艾杰尔科技有限公司;
十八烷基硅烷键合硅胶(C18):40~60 μm,博纳艾杰尔科技有限公司;
针筒式微孔滤膜:孔径0.22 μm,材质为英国尼龙66(有机系),天津市津腾实验设备有限公司;
超纯水:电阻率≥18.2 MΩ·cm(25 ℃),实验室自制。
1.2 主要试验仪器
三重四级杆串联质谱仪:Triple Quad 5500,美国AB SCIEX公司;
液相色谱仪:LC-30A型,日本岛津公司;
电子分析天平:XS204型,梅特勒—托利多仪器(上海)有限公司;
电子分析天平:ME2002E型,梅特勒—托利多仪器(上海)有限公司;
高速离心机:Avanti JXN-30型,美国贝克曼库尔特有限公司;
翻转式混匀器:Trayster Digital型,德国IKA公司;
氮吹仪:N-EVAP24型,美国Organomation公司;
涡旋混合器:基本型,美国Talboys公司。
1.3 标准对照品的配制
1.3.1 标准对照品储备溶液 分别准确称取灭草松、6-羟基灭草松、8-羟基灭草松标准品10.00 mg,根据标准品的溶解性和测定需要分别用甲醇溶解并定容至100 mL,配制成浓度为100 μg/mL标准对照品储备溶液,-20 ℃保存。
1.3.2 混合标准对照品中间溶液 分别准确吸取1.00 mL 灭草松、6-羟基灭草松、8-羟基灭草松标准储备溶液于同一个100 mL容量瓶中,用甲醇稀释并定容,即得1.0 μg/mL 混合标准中间溶液,4 ℃保存。
1.3.3 基质混合标准对照品工作液 取经净化后的空白基质溶液1 mL(取样量与相应的试样处理取样量相同),氮气吹干,用1.00 mL相应质量浓度的混合标准对照品溶液复溶,过微孔滤膜,滤液待测。基质混合标准对照品工作溶液应保证现用现配。
1.4 样品前处理
1.4.1 提取 准确称取已研磨的大米或小麦粉5.00 g于50 mL带盖聚乙烯离心管中,先加入10 mL超纯水,充分涡旋均匀,静置30 min。再同时加入10 mL乙腈和0.5 mL 6 mol/L HCl溶液,加入5 g无水硫酸钠,剧烈振荡涡旋2 min,翻转提取20 min,4 000 r/min离心5 min,将上清液置于50 mL离心管中,待净化。
1.4.2 净化 吸取2 mL上清液加入至含200 mg PSA、100 mg C18粉末和200 mg无水硫酸镁的15 mL塑料离心管中,涡旋混合2 min,4 000 r/min 离心5 min,上清液过0.22 μm有机滤膜,高效液相色谱—串联/质谱仪待测。
1.5 液相色谱参考条件的确定
对比不同的色谱柱与不同的有机相与水相溶液配比(见表1),通过比较色谱行为,选择合适的色谱柱和流动相。对梯度洗脱条件进行优化,并设置和调节仪器的进样条件,以达到分离度高、峰型优和响应最佳的色谱行为,从而确定液相色谱条件。
1.6 质谱参数条件的确定
1.6.1 分子离子峰的确定 配制一定浓度的标准溶液,采用全自动进样,扫描范围为200~300 Da,运用Q1 MSFull Scan模式正、负离子模式进行扫描,得到对应的质谱总离子流图,并通过总离子流图确定化合物的分子离子峰。
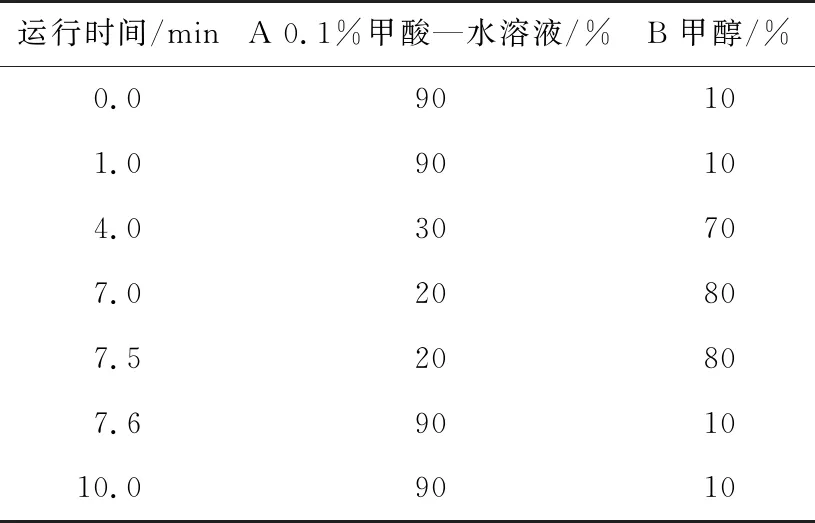
表1 液相色谱梯度洗脱程序Table 1 Liquid chromatography gradient elution procedure
1.6.2 二级质谱扫描 选择Product ion扫描模式,扫描范围为50~300 Da,输入分子离子质核比数值,施加碰撞电压进行二级质谱扫描。
1.6.3 化合物质谱条件的优化 对目标化合物质谱条件的优化项目包括电喷雾电压、离子源温度、气帘气压力、雾化气压力、辅助气压力、去簇电压、碰撞气能量。选择二级质谱中相对响应较高的子离子进行MRM质谱条件的优化,记录最优的去簇电压和碰撞气能量,生成化合物质谱条件。
1.7 提取条件和净化方式的优化
比较不同的有机试剂以及pH值对提取过程的影响,以提取效率为指标,确定提取条件;对PSA、C18、MgSO4用量进行正交试验设计,以灭草松、6-羟基灭草松、8-羟基灭草松含量的检出值为参考指标,通过极差分析确定最终优化添加量,从而确定合适的净化方式。
1.8 方法学验证
1.8.1 标准曲线绘制及样品的测定 取1.3.3中的混合标准系列对照品工作液,按优化后液相色谱—串联质谱条件进行进样分析,记录对应定量离子的峰面积,以浓度对峰面积进行绘制和拟合标准曲线,得到对应的回归方程和相关系数。对于市售大米及面粉样品,按1.4前处理后,经液相色谱—串联质谱检测,以保留时间和相应的定量、定性离子对定性,定量离子的峰面积外标法定量。
1.8.2 准确度和精密度 准确度用样品加标回收率衡量,精密度用相对标准偏差的百分数表示。不同的基质样品进行3个加标浓度(低、中、高)的添加,分别测定回收率并计算相对标准偏差的百分数(RSD%)。
1.8.3 最低定量限(LOQ) LOQ由10倍信噪比得出(S/N=10)。
1.9 数据处理
相关质谱数据由Analyst分析软件采集,MultiQuant 定量软件分析数据。数据汇总后,采用Microsoft Office Excel进行数据处理。
2 结果与讨论
2.1 色谱条件的确定
灭草松、6-羟基灭草松、8-羟基灭草松的极性相对较弱,在普通C18色谱柱都能较好地保留。6-羟基灭草松和8羟基灭草松为同分异构体,色谱柱以及流动相的选择既要保证重叠峰有最佳分离度同时也要保证色谱峰的峰型及质谱响应,试验选择Waters ACQUITY UPLC BEH C18色谱柱(100 mm×2.1 mm,1.7 μm),较Waters ACQUITY UPLC BEH C18色谱柱(50 mm×2.1 mm,1.7 μm)能达到更好的分离度。试验表明,0.1%甲酸水—甲醇流动相体系不仅可以改善峰型,且在7 min内可以达到较好的基线分离,更有利于化合物的定性和定量分析。故确定最优色谱条件:色谱柱为Waters ACQUITY UPLC BEH C18色谱柱(100 mm ×2.1 mm,1.7 μm);色谱柱温度40 ℃;流动相A为0.1%甲酸—水溶液,B为甲醇,梯度洗脱;流动相流速0.3 mL/min;进样量5 μL。最优色谱质谱条件下各待测物质的MRM色谱图见图2。由图2可知,3种化合物峰型良好,分离度高,无干扰。
2.2 质谱条件的确定
试验表明,ESI-模式下,3种化合物的分子离子[M-H]-均最为理想;然后分别以目标化合物的 [M-H]-作为母离子,施加碰撞能量将其打碎,同时进行子离子扫描,根据二级碎片离子扫描质谱图及可能的断裂规律,选择干扰较少、相对丰度较高的两个子离子,并在负电离模式下以多反应监测(MRM)方式优化碰撞电压(CE)和去簇电压(DP),灭草松和6-羟基灭草松及8-羟基灭草松的二级质谱图见图3。确定扫描方式为负离子扫描,气帘气压力为275 kPa,电喷雾电压为-4 500 V(负离子模式),离子源温度为450 ℃,雾化气压力为345 kPa,辅助气压力为380 kPa,检测方式为多反应监测(MRM),相应的质谱条件见表2。
2.3 样品提取溶剂优化
农药残留测定常用的萃取溶剂有甲醇、乙腈、丙酮、乙酸乙酯、正己烷等[19]。根据相似相溶原理,提取溶剂的极性和样品基质的类型决定目标物质的提取效率[20]。大米和面粉作为粮谷类产品进行农药残留测定前需先加入一定量的水进行浸润和溶胀,使提取试剂与样品充分接触互溶从而利于提取。试验表明,甲醇提取液经盐析也无法与水相分离;丙酮与水互溶,加大后续提取液的净化难度;正己烷和乙酸乙酯的提取率不高,故选择乙腈作为提取液,加入无水硫酸钠进行盐析。

图2 灭草松及其代谢物的MRM色谱图Figure 2 MRM chromatograms of bentazone,6-hydro- xymetazone,8-hydroxymetazone

图3 灭草松及其代谢物的二级质谱图Figure 3 Secondary mass spectra of bentazone,6-hydroxymetazone,and 8-hydroxymetazone
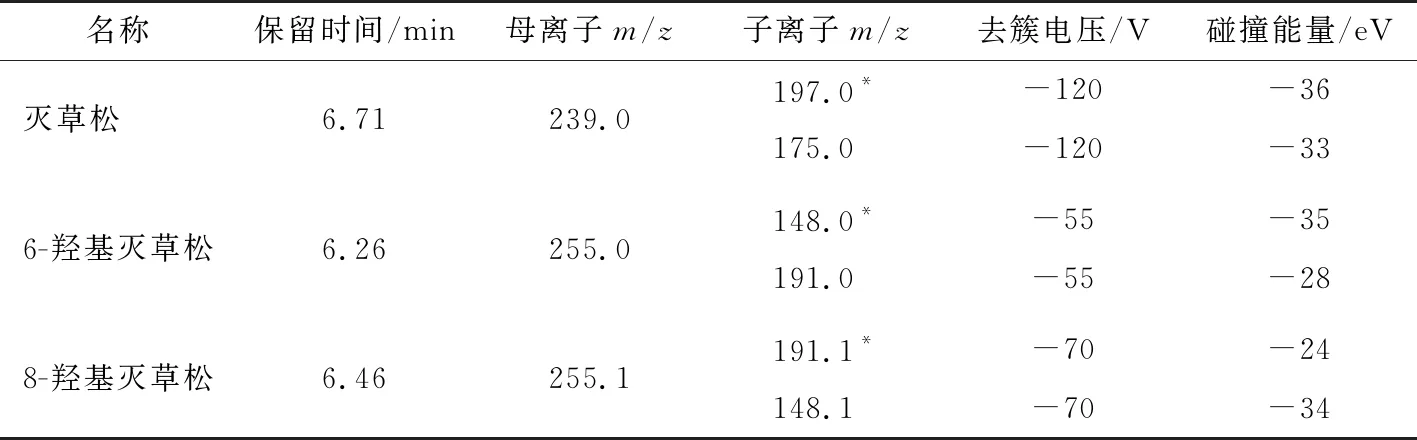
表2 灭草松及其代谢物的质谱条件†Table 2 The main reference mass spectrometry conditions of bentazone and its metabolites
灭草松及其代谢物的pKa值较小,降低溶液的pH值能有效地使目标物由离子状态转为分子形式析出,从而使其在有机溶剂中的分配效率升高[21]。试验采取直接加入酸的方式调节pH值,各目标化合物的加标回收率见表3。由表3可知,加酸后目标化合物的回收率明显高于未加酸的;加入盐酸后的提取效果要明显高于甲酸、乙酸等有机酸,且当HCl添加量为0.5 mL时,灭草松、6-羟基灭草松、8-羟基灭草松的回收率均高于其他添加情况。因此,提取过程中加入0.5 mL 6 mol/L的盐酸溶液有助于提高提取率,从而提高过程回收率。
2.4 样品净化步骤优化
研究[22]表明,石墨化碳黑粉、Al2O3粉对除草剂有较强的吸附,不建议作为净化吸附剂。净化小柱HLB按通用净化过程,经活化、上样、淋洗、洗脱处理后,灭草松及其代谢物回收率均较低;直接提取后稀释有基质效应且对仪器有一定程度损伤。故在最优条件提取后的溶液中加入PSA、C18、MgSO4进行净化,综合参照文献[23-25]和国家标准[26-27]的通用用量添加,以灭草松、6-羟基灭草松、8-羟基灭草松含量的检出值为参考指标设计正交试验。正交试验因素水平见表4,试验结果见表5、6。
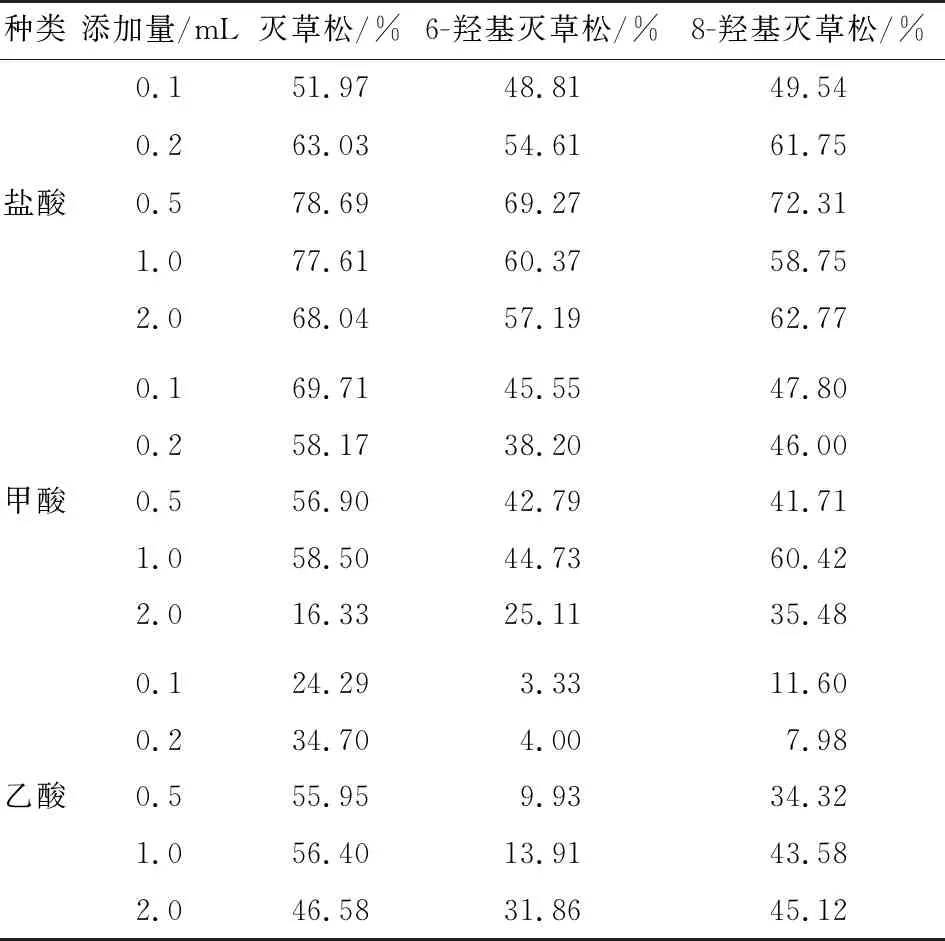
表3 酸添加量对灭草松及其代谢物提取率的影响Table 3 Effect of acid addition on the extraction rate of Bentazone and its metabolites
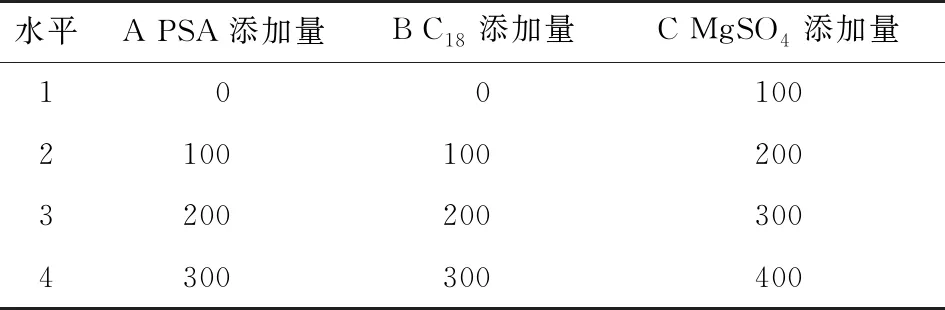
表4 正交试验因素水平表Table 4 Factor level table of orthogonal test mg
由表6可知,灭草松的最优净化方式为PSA 300 mg、C18200 mg、MgSO4200 mg;6-羟基灭草松的最优净化方式为PSA 200 mg、C18100 mg、MgSO4200 mg;8-羟基灭草松的最优净化方式为PSA 200 mg、C18100 mg、MgSO4400 mg。当PSA添加量分别为200,300 mg 时,灭草松含量无显著差异;当C18添加量分别为100,200 mg时,灭草松含量变化不显著;MgSO4作为吸水剂,对灭草松及其代谢物含量影响不显著。综合考虑,选择更少的添加量试剂:PSA 200 mg、C18100 mg、MgSO4200 mg。
2.5 基质效应
试验使用基质匹配标准曲线与溶剂标准曲线的斜率之比评估基质效应作用,即当该比率为0.8~1.2时,为弱的基质效应;当该比率<0.8时,为基质抑制作用;当该比率>1.2时,为基质增强作用[28]。由表7可知,灭草松、6-羟基灭草松、8-羟基灭草松的基质匹配标准曲线与溶剂标准曲线斜率比值分别为1.06,1.42,1.82,说明灭草松具有较弱的基质增强效应,对结果的准确性影响不大,而其代谢产物具有较强的基质作用,即基质效应对结果产生的干扰较大。为了消除基质增强效应对测定结果的影响,试验拟使用基质匹配标准曲线的方式进行校准,以保证数据的准确性。
2.6 方法的线性范围与检出限
由表8可知,当样品浓度为0.1~100.0 μg/kg时,灭草松、6-羟基灭草松及8-羟基灭草松基质匹配标准曲线线性关系良好,相关系数均>0.995,LOQ均能满足GB 2763—2019中灭草松及其代谢物临时限量检测的要求。
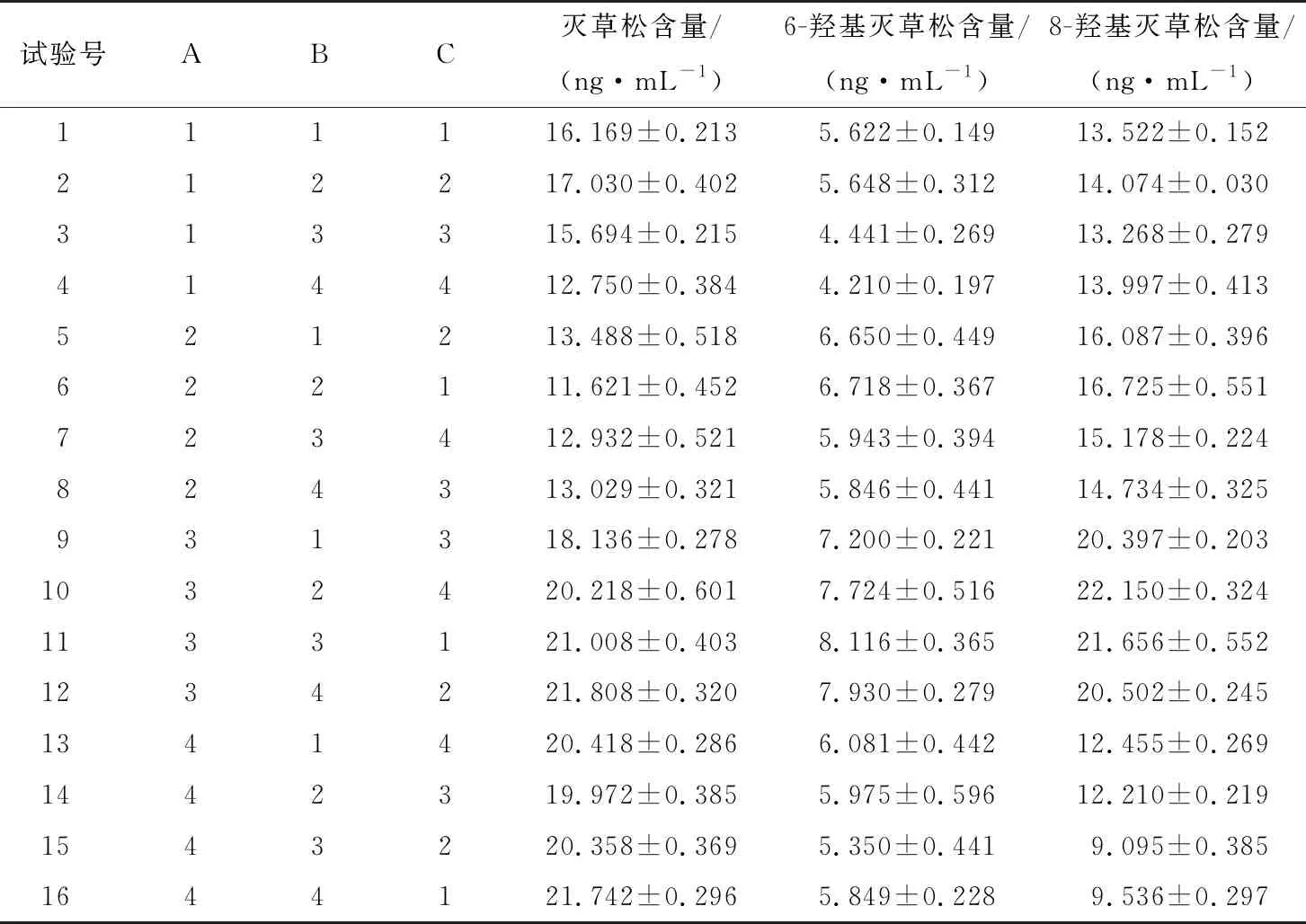
表5 正交试验结果Table 5 Orthogonal test results
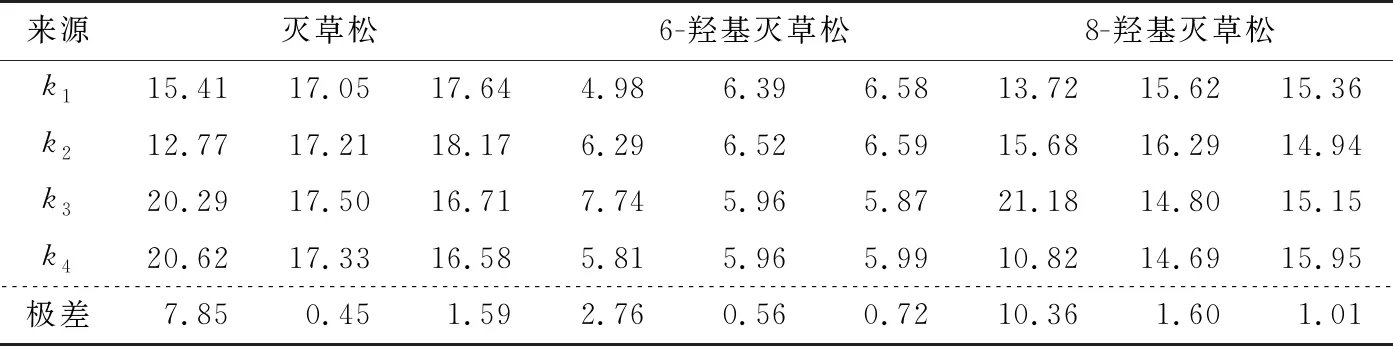
表6 极差分析Table 6 Range analysis of each parameter

表7 灭草松及其代谢物的基质效应Table 7 Matrix effects of Bentazone and its metabolites
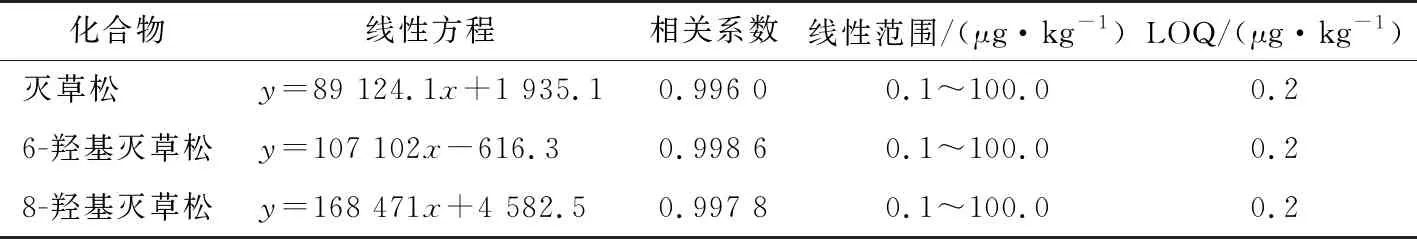
表8 灭草松及其代谢物的线性方程、相关系数、线性范围和定量限Table 8 Linear equation,correlation coefficient,linear range and limit of quantification of bentazone and its metabolites
2.7 方法的精密度与准确度
由表9可知,灭草松、6-羟基灭草松及8-羟基灭草松的回收率为89.6%~106.9%,相对标准偏差为1.4%~11.7%,回收率和相对标准偏差均符合GB/T 27404—2008实验室质量控制规范,能够满足灭草松及其代谢物的残留分析要求。
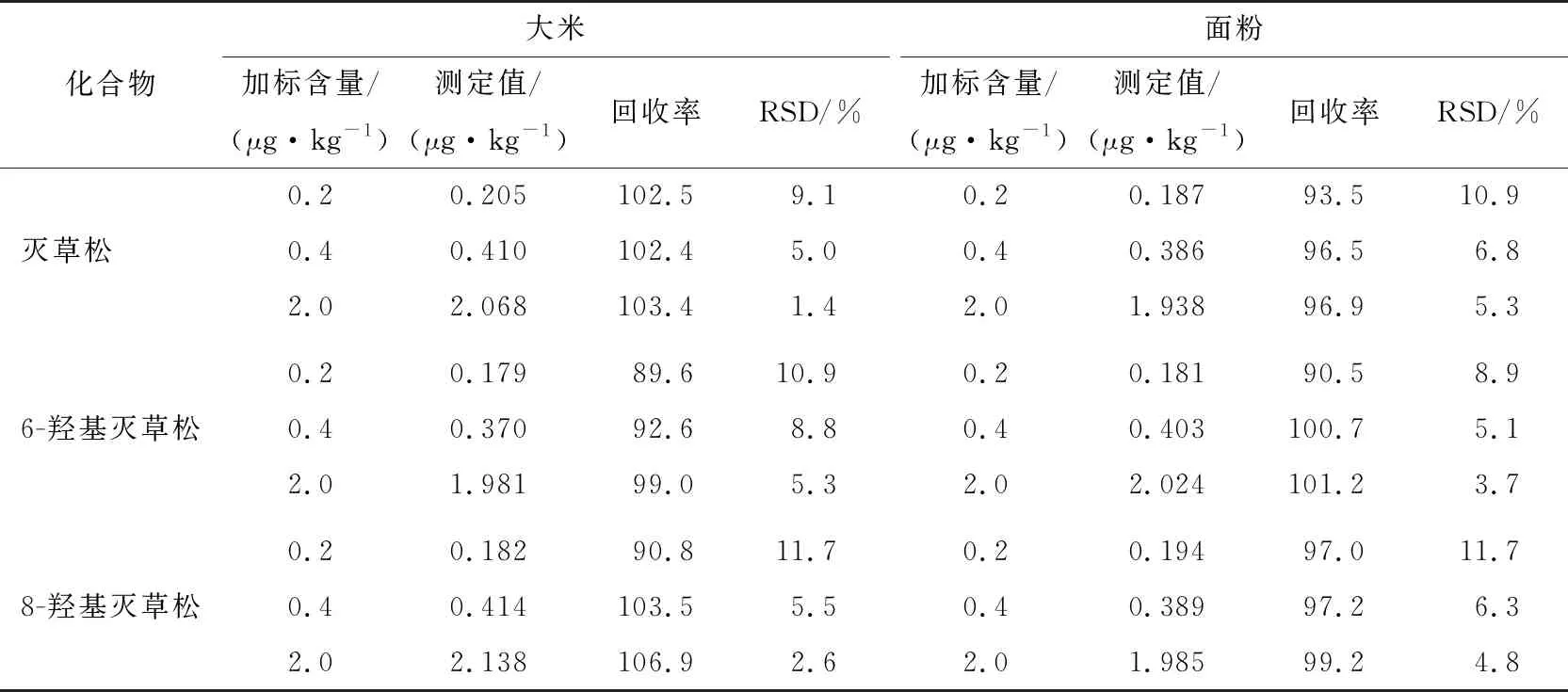
表9 大米和面粉中灭草松及其代谢物的加标回收率及精密度Table 9 Recoveries rate and precision experiment (RSDs) of Bentazone and its metabolites in rice and flour (n=6)
2.8 实际样品的测定
按试验检测方法对20批次武汉市各大农贸市场的市售大米和面粉样品进行提取检测。结果表明,各市售大米和面粉样品均未检出灭草松及其代谢物残留。
3 结论
采用优化的QuEChERS净化方法,通过液相色谱—串联质谱法测定大米和面粉中灭草松及其代谢物残留量。结果表明,该检测方法的可操作性强、步骤简单快捷、色谱峰在7 min内分离效果好,准确度和精密度良好,能适用于实际样品检测,效果理想。灭草松及其代谢物在0.1~100.0 μg/kg浓度范围内线性关系良好,方法定量限为0.2 μg/kg,平均加标回收率为89.6%~106.9%,相对标准偏差为1.4%~11.7%。试验仅针对粮谷类基质进行了研究,对于油料作物和油脂等基质相对复杂的农产品的前处理方法也亟待开发。后续应加大对不同种类、地域的农产品监测,完善其他不同种类基质的农产品检测方法,为最大限量值地制定和法定标准方法的生成提供理论依据。
猜你喜欢
现代临床医学(2022年4期)2022-09-29
中山大学学报(自然科学版)(中英文)(2022年4期)2022-08-05
当代水产(2022年4期)2022-06-05
口腔护理用品工业(2021年4期)2021-11-02
环境保护与循环经济(2021年7期)2021-11-02
食品安全导刊(2021年20期)2021-08-30
昆明医科大学学报(2021年1期)2021-02-07
农药科学与管理(2019年5期)2019-08-13
人间(2015年11期)2016-01-09
数理化学习·高一二版(2009年1期)2009-03-19
