
非甾体抗炎药酮洛芬的合成
2021-02-05 08:04金文斐余刘东李泓江袁明伟李宏利江登榜袁明龙
云南大学学报(自然科学版) 2021年1期
金文斐,余刘东,李泓江,袁明伟,李宏利,江登榜,袁明龙,蒋 琳
(云南民族大学 化学与环境学院,生物基材料绿色制备技术国家地方联合工程研究中心,云南 昆明 650504)
酮洛芬(ketoprofen),又名酮基布洛芬,化学名为α-甲基-3-苯甲酰基苯乙酸,结构如图1所示,属2-芳基丙酸类非甾体抗炎药物. 该化合物分子中含有一个手性中心,产生一对对映异构体,其中,右旋酮洛芬作为抗炎抗风湿的药效成分,而左旋酮洛芬几乎无药理活性,临床以外消旋体供药为主[1]. 酮洛芬由法国Rhone-Poulenc公司研发并于1973年首次在法国上市,作用机制主要是通过抑制体内环氧合酶、脂氧化酶的生物活性,减少致炎物质前列腺素、白三烯的合成,从而产生良好的解热、镇痛以及抗炎作用. 酮洛芬临床上适用于治疗各类关节炎和术后、癌症疼痛,与同类药物布洛芬、萘普生等相比,具有药效强、剂量小、耐受性好、口服易吸收等优点[2].
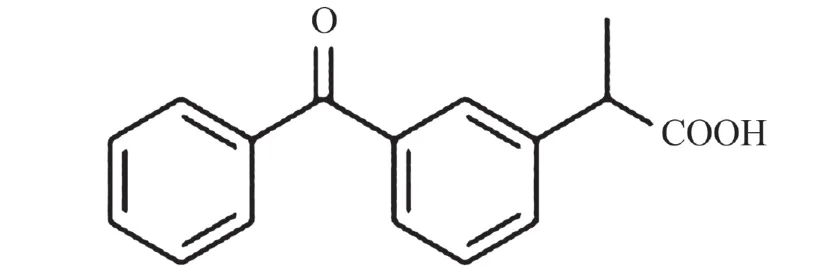
图 1 酮洛芬的化学结构Fig. 1 Chemical structure of ketoprofen
酮洛芬的化学结构包含二苯甲酮和α-取代的丙酸侧链两部分,已报道的合成方法多数是以间位甲基或卤素(Cl, Br)取代的苯甲酸为起始原料,经傅克反应合成二苯甲酮片段,再通过苯环间位的甲基或卤素的化学转化拼合侧链部分(图2). 如:苯乙腈法经3-甲基二苯甲酮的苄位卤代、氰基取代、甲基化、水解得到(路线1)[3];3-苯甲酰基苯乙酮法从3-溴二苯甲酮经格氏反应、Darzen反应、氧化得到(路线2)[4];丙二酸二乙酯法从3-溴二苯甲酮经偶联、苄位甲基化、水解、脱羧反应得到(路线3)[5].此外,文献报道的合成方法还包括1,2-芳基迁移法[6]、苯胺法[7]、苯乙烯法[8]、苯乙酮法[9],等. 迄今为止,虽然酮洛芬的合成路线较多[10, 11],但仍然存在各种缺点,例如:苯乙腈法需要用剧毒的氰化试剂,另外,苄位甲基化时易形成单取代和二取代的混合物,使得后续难分离[4, 12];3-苯甲酰基苯乙酮法用格氏反应合成3-乙酰基二苯甲酮需要预先保护底物中的羰基,且格氏反应条件严苛,操作繁琐;1,2-芳基迁移法采用四乙酸铅作重排试剂,易造成药物中重金属残留;苯胺法、苯乙烯法用氧化反应生成二苯甲酮的羰基,存在潜在的火灾爆炸危险性,因此,传统工艺面临创新和优化的压力.
综合考虑已报道方法的优、缺点,我们选择对丙二酸二乙酯法进行以下改进和优化:①采用碘化亚铜为催化剂,2-苯基苯酚为配体,通过偶联反应合成2-(3-苯甲酰苯基)丙二酸二乙酯(2);②碳酸钾作碱,采用相转移反应条件实现化合物2的苄位甲基化;③采用Krapcho反应脱酯基、再水解生成酮洛芬中α-取代的丙酸侧链. 设计的合成路线如图3所示.
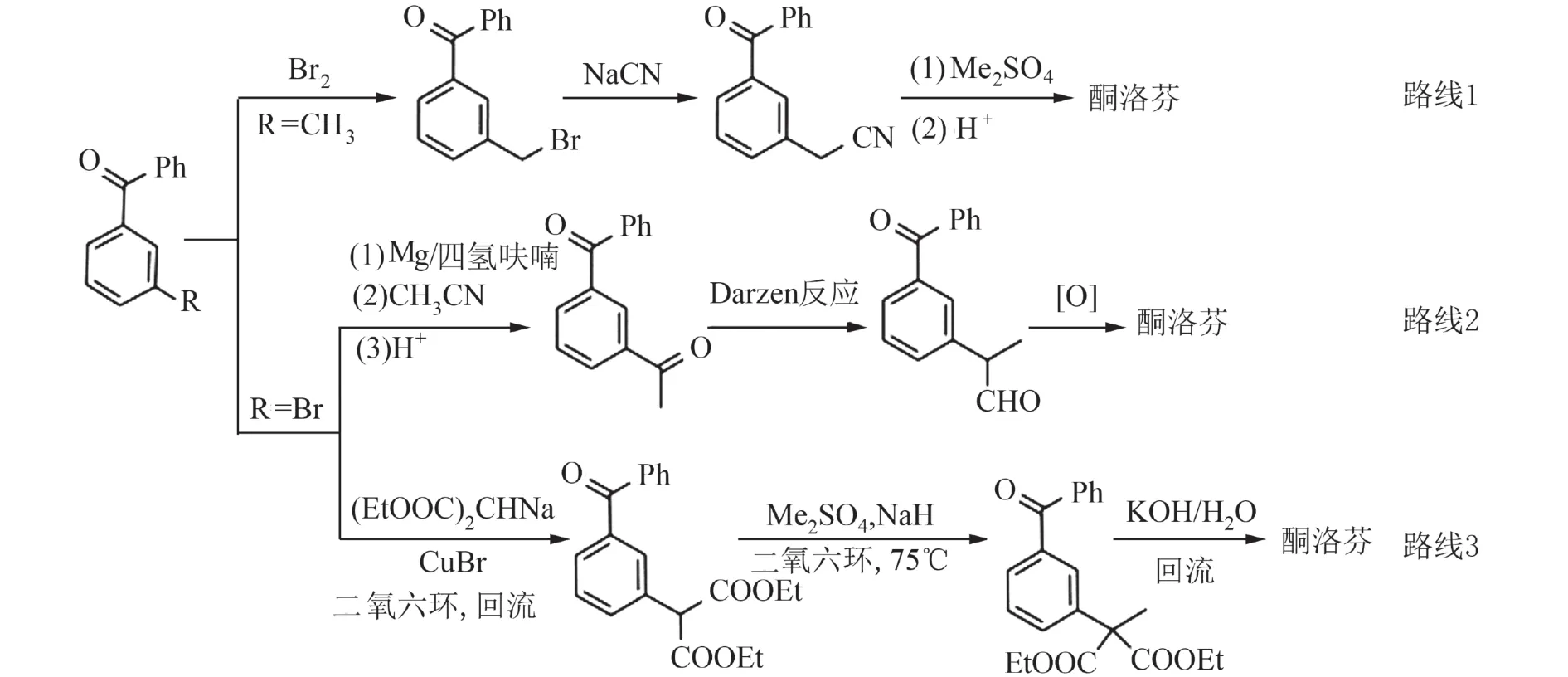
图 2 代表性的酮洛芬合成路线Fig. 2 Representative synthetic routes of ketoprofen
1 实验部分
1.1 仪器与试剂Optimelt全自动熔点仪(美国Stanford 大学研究所);BrukerAvance-II 400 MHz型核磁共振仪(德国Bruker公司);IS10型傅里叶变换红外光谱仪(美国Nicolet公司);LTQ-Orbitrap XL 高分辨质谱仪(美国 Thermo Fisher Scientific公司);Finnigan LCQDECA型液质联用仪 (美国Thermo Finnigan 公司).
3-碘苯甲酸、丙二酸二乙酯、碳酸铯、碘化亚铜、氯化钠等所用试剂均为市售分析纯或化学纯,购买自上海泰坦、阿拉丁等生化科技股份有限公司.
1.2 合成方法
1.2.1 3-碘二苯甲酮(1)的合成 3-碘苯甲酸(100.0 g, 0.40 mol)溶于 200 mL 二氯甲烷 (DCM),室温下加入氯化亚砜 (34.8 mL, 0.48 mol)和 0.5 mL N,N-二甲基甲酰胺,回流反应 4 h,减压浓缩,残余物溶于 100 mL 苯,于 0 °C 下分批加入三氯化铝(186.6 g, 1.4 mol),升温至 60 °C 反应 5 h,将反应液倒入100 g碎冰和60 mL浓盐酸的混合物中进行淬灭,分离有机相,水相用适量乙酸乙酯分2次萃取,合并有机相,用饱和碳酸氢钠洗至中性,再用饱和食盐水洗1次,无水硫酸钠干燥,减压浓缩溶剂,残余物用甲基叔丁基醚和乙醇混合溶剂重结晶,得淡黄色固体 107.8 g,为 3-碘二苯甲酮(1),产率:88%. m.p. 41~42 °C(文献值[13]41~42 °C);1H NMR(400 MHz, CDCl3)δ: 8.13 (d,J= 4.0 Hz, 1H), 7.91 (d,J= 8.0 Hz, 1H), 7.78 (d,J= 8.0 Hz, 2H), 7.74 (d,J=8.0 Hz, 1H), 7.63~7.59 (m, 1H), 7.51~7.48 (m, 2H),7.24~7.20 (m, 1H);13C NMR (100 MHz, CDCl3)δ:195.2, 141.3, 139.6, 138.7, 137.0, 133.0, 130.2, 130.1,129.3, 128.6, 94.2.
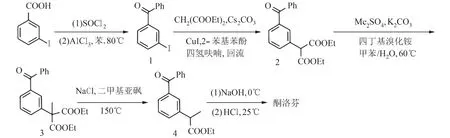
图 3 酮洛芬的合成路线设计Fig. 3 Synthetic route design of ketoprofen
1.2.2 2-(3-苯甲酰苯基)丙二酸二乙酯(2)的合成 500 mL圆底烧瓶中依次加入碘化亚铜(3.3 g, 17.5 mmol),2-苯基苯酚 (6.0 g, 35.0 mmol),碳酸铯 (228.1 g, 0.70 mol),氩气保护下,依次加入 100 mL无水四氢呋喃、化合物 1(107.8 g, 0.35 mol)和丙二酸二乙酯 (215.2 mL, 0.70 mol),于 70 °C 避光反应24 h,反应液冷却至室温,加入 100 mL 饱和氯化铵淬灭反应,乙酸乙酯提取3次,合并有机相,无水硫酸钠干燥,减压浓缩溶剂,得淡黄色液体102.1 g,为2-(3-苯甲酰苯基)丙二酸二乙酯(2)的粗品,可不经纯化直接用于下步反应. 化合物2的结构表征数据与文献 [14]报道一致.1H NMR (400 MHz,CDCl3)δ: 7.82~ 7.76 (m, 4H), 7.68 (d,J= 8.0 Hz,1H), 7.61~7.57 (m, 1H), 7.51~7.47 (m, 3H), 4.69 (s,1H), 4.26~4.18 (m, 4H), 1.26 (t,J= 8.0 Hz, 6H);13C NMR (100 MHz, CDCl3)δ: 196.3, 167.9, 137.9,137.5, 133.4, 133.2, 132.7, 131.2, 130.2, 130.1, 128.7,128.5, 62.2, 57.9, 14.1.
1.2.3 2-甲基-2-(3-苯甲酰苯基)丙二酸二乙酯(3)的合成 100 mL 水中加入碳酸钾 (103.7 g, 0.75 mol)和四丁基溴化铵 (9.7 g, 0.03 mol),转移至上步反应所得化合物 2(102.1 g, 0.30 mol, 作纯品计)的 200 mL甲苯溶液中. 加热至60 °C,搅拌下向反应瓶中缓慢滴加硫酸二甲酯 (42.6 mL, 0.45 mol)的 50 mL 甲苯稀释液,约 40 min 滴加完,60 °C 反应 8 h,反应液冷却至室温,分离有机相,用10%氨水洗3次,饱和食盐水洗至中性,无水硫酸钠干燥,减压浓缩溶剂,得淡黄色液体 95.7 g,为 2-甲基-2-(3-苯甲酰苯基)丙二酸二乙酯(3)的粗品,可不经纯化直接用于下步反应.1H NMR (400 MHz, CDCl3)δ: 7.83 (d,J=4.0 Hz, 1H), 7.77 (d,J= 8.0 Hz, 2H), 7.71 (d,J= 8.0 Hz, 1H), 7.61 (d,J= 8.0 Hz, 1H), 7.56~7.53 (m, 1H),7.46~7.42 (m, 3H), 4.24~4.16 (m, 4H), 1.87 (s, 3H),1.21 (t,J= 8.0 Hz, 6H);13C NMR (100 MHz, CDCl3)δ: 196.2, 171.0, 138.6, 137.4, 137.3, 132.5, 131.7,130.0, 129.3, 129.2, 128.3, 128.1, 61.9, 58.7, 22.2,13.9; IR (KBr)ν: 2 981, 1 734, 1 661, 1 449, 1 284,1 179, 1 078, 721 cm−1; ESI-HRMSm/z: C21H22O5[M+Na]+计算值 377.136 5, 测定值 377.1362.
1.2.4 α-甲基-(3-苯甲酰基)苯乙酸乙酯(4)的合成 上步反应所得化合物 3 (95.7 g, 0.27 mol,作纯品计)溶于80 mL二甲基亚砜,加入氯化钠(31.5 g,0.54 mol)和水 (19.4 mL, 1.08 mol), 150 °C 反应约6 h,冷却至室温,加入 100 mL 水,乙酸乙酯提取3次,合并有机相,再用适量水洗3次,饱和食盐水洗1次,无水硫酸钠干燥,减压浓缩溶剂,残余物经硅胶快速柱层析纯化(石油醚-乙酸乙酯 体积比15:1),得淡黄色液体 67.8 g,为 α-甲基-(3-苯甲酰基)苯乙酸乙酯(4),产率:69% (以化合物 1 摩尔数计,共计3步反应),化合物4的结构表征数据与文献 [9]报道一致.1H NMR (400 MHz, CDCl3)δ: 7.64(d,J= 8.0 Hz, 2H), 7.61 (d,J= 4.0 Hz, 1H), 7.52 (d,J= 4.0 Hz, 1H), 7.44~ 7.39 (m, 2H), 7.34~ 7.28(m,3H), 4.02~3.93 (m, 2H), 3.63 (q,J= 8.0 Hz, 1H), 1.37(d,J= 8.0 Hz, 3H), 1.06 (t,J= 4.0 Hz, 3H);13C NMR(100 MHz, CDCl3)δ: 196.5, 174.1, 141.0, 137.9,137.5, 132.5, 131.6, 130.1, 129.2, 129.0, 128.6, 128.3,61.0, 45.4, 18.6, 14.2.
1.2.5 酮洛芬的合成 化合物 4(67.8 g, 0.24 mol)溶于 100 mL 乙醇,将 NaOH(10.4 g, 0.26 mol)溶于10 mL 乙醇,0 °C 下滴加到反应液中,10 min 滴完,0 °C 反应约 30 min,TLC 监测化合物 4 转化情况.转化完全后向反应液中滴加浓度为2 mol/L的稀盐酸,调节反应液pH至强酸性,加入乙酸乙酯和水进行萃取,水相再用乙酸乙酯提取2次,合并有机相,食盐水洗1次,无水硫酸钠干燥,减压浓缩溶剂,待残余物固化后用甲基叔丁基醚和乙醇混合溶剂重结晶,得白色固体58.5 g,为目标产物酮洛芬,产率:96%. m.p. 93~94 °C(文献[11]: 93.7~95.2 °C). 目标产物的结构表征数据与文献[15]报道一致.1H NMR (400 MHz, CDCl3)δ: 7.81~7.78 (m, 3H), 7.69(d,J= 8.0 Hz, 1H), 7.61~7.56 (m, 2H), 7.50~ 7.43(m, 3H), 3.83 (q,J= 8.0 Hz, 1H), 1.56 (d,J= 8.0 Hz,3H);13C NMR (100 MHz, CDCl3)δ: 196.0, 179.4,141.6, 138.7, 137.6, 133.4, 131.8, 130.2, 129.5, 128.8,127.6, 44.2, 18.3; ESI-MSm/z: 255 [M+H]+.
2 讨论
2.1 偶联反应合成 2-(3-苯甲酰苯基)丙二酸二乙酯(2)丙二酸二乙酯法以亚铜盐催化3-溴二苯甲酮和丙二酸二乙酯偶联,生成中间体2-(3-苯甲酰苯基)丙二酸二乙酯(2). 传统方法中溴化亚铜用量多达1.5倍量比,而丙二酸二乙酯用量为2.5倍量比且需要预制成钠盐. 改进的合成工艺采用活性更强的3-碘二苯甲酮为原料,在碘化亚铜为催化剂,2-苯基苯酚为配体,碳酸铯作碱的催化体系中反应,大大减少了亚铜盐和丙二酸二乙酯的用量(分别为0.025倍量比和1倍量比),且丙二酸二乙酯不需要预先成盐,简化合成步骤的同时也节约了反应原料.
2.2 甲基化反应合成 2-甲基-2-(3-苯甲酰苯基)丙二酸二乙酯(3)由于双酯基的活化效应,2-(3-苯甲酰苯基)丙二酸二乙酯(2)的苄位具有较高的反应活性,改进的合成方法采用相转移反应实现苄位甲基化,用碳酸钾代替传统方法中的氢化钠,反应条件温和,操作更安全.
2.3 酮洛芬侧链的合成传统方法从 2-甲基-2-(3-苯甲酰苯基)丙二酸二乙酯(3)合成酮洛芬侧链时采用先水解,再加热脱羧的顺序,水解生成的羧酸根高温下易和二苯甲酮的羰基缩合. 改进的合成方法采用Krapcho反应脱酯基、再水解的顺序. 由于水解生成的羧酸根只存在于低温下,只要水解完全后及时酸化,即可很好地避免缩合副反应的发生.
3 结论
以3-碘苯甲酸为起始原料,经过傅克酰基化、催化偶联、苄位甲基化、Krapcho脱酯基、酯水解共5步反应合成非甾体抗炎药酮洛芬,总产率为58%. 优化后的合成方法与传统方法相比,具有化学选择性好、操作简便、反应条件温和、原子经济性较高的优点,具有较好的工业化应用前景.
猜你喜欢
长江大学学报(自科版)(2021年3期)2021-06-01
探索科学(学术版)(2020年4期)2021-01-18
理化检验-化学分册(2020年5期)2020-06-15
电脑报(2020年10期)2020-04-28
山东化工(2016年4期)2016-09-05
灾害医学与救援(电子版)(2016年2期)2016-03-11
当代医药论丛(2016年3期)2016-03-08
医学研究杂志(2015年3期)2015-06-10
中国当代医药(2015年8期)2015-03-01
中国卫生标准管理(2015年3期)2015-01-27
