
高压制备纽莫康定B0的中试研究
2021-03-18 10:13曾凡洲朱兵峰刘志钰王钦超张贵民魏荣省
发酵科技通讯 2021年1期
曾凡洲,朱兵峰,刘志钰,王钦超,张贵民,魏荣省
(1.鲁南新时代生物技术有限公司,山东 临沂 273400;2.鲁南制药集团股份有限公司,山东 临沂 273400;3.哺乳动物细胞高效表达国家工程实验室,山东 临沂 273400)
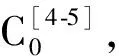
色谱分离法在化合物的分离中应用较为广泛,且色谱纯度、回收率和分离效率等方面远远优于传统的制备方法[7-9],其中色谱纯度通过面积归一化法得到。相比于中低压色谱制备,高压色谱制备效率高、柱效高。笔者以实验室制得的纽莫康定B0粗品为原料,通过高压制备对纽莫康定B0进行了放大中试研究,开发出了一种生产周期短、成本低和色谱纯度高的制备工艺。
1 仪器与试药
1.1 仪 器
U3000高效液相色谱仪,赛默飞Thermo公司;Cs-prep150工业制备色谱系统,汉邦科技;R-1001N型旋转蒸发仪,郑州长城科工贸有限公司;SHB-B95型循环水式多用真空泵,郑州长城科工贸有限公司;DHJK-2020低温冷却循环泵,郑州科泰实验设备有限公司;SCIENTZ-12ND冷冻干燥机,宁波新芝生物科技股份有限公司。
1.2 试 药
工业级二氯甲烷,山东金岭化工股份有限公司;工业甲醇,兖矿国宏化工有限责任公司;改性硅胶,苏州纳微科技股份有限公司;纽莫康定B0粗品,鲁南新时代生物技术有限公司提供,质量分数81.20%;纽莫康定B0对照品,大同市矿区风华生物科技有限公司,质量分数95.56%,批号16001;纽莫康定C0对照品,大同市矿区风华生物科技有限公司赠与,质量分数95.11%,批号16002;乙腈为色谱纯,其他试剂均为分析纯,水为纯化水。
2 实 验
2.1 HPLC(高效液相色谱)法测定B0
纽莫康定B0检测方法[10]:Thermo Hypersil GOLD C18,4.6 mm×250 mm,5 μm;以V(乙腈)∶V(水)=45∶55为流动相,等度洗脱;检测波长210 nm,柱温40 ℃,进样量10 μL,流量1 mL/min,运行时间45 min。以纽莫康定B0对照品进行外标法定量。纽莫康定B0对照品溶液的配制:取纽莫康定B0对照品约20 mg,精密称定,置于50 mL容量瓶中,加乙醇超声溶解后,稀释至刻度,摇匀,制得质量浓度为400 mg/L的纽莫康定B0对照品溶液。供试品溶液配制:对制备液用氮气吹干,加乙醇振摇使溶解并稀释至质量浓度约1 000 mg/L,摇匀,用0.22 μm有机滤头过滤。纽莫康定B0的结构式为
2.2 HPLC(高效液相色谱)法测定C0
检测方法为公司自行开发,线性梯度洗脱程序如表1所示。Welch Ultimate HILIC Amide柱,4.6 mm×250 mm,3.5 μm;波长220 nm,流量1.0 mL/min,进样量10 μL,柱温40 ℃,流动相A为水,流动相B为乙腈。
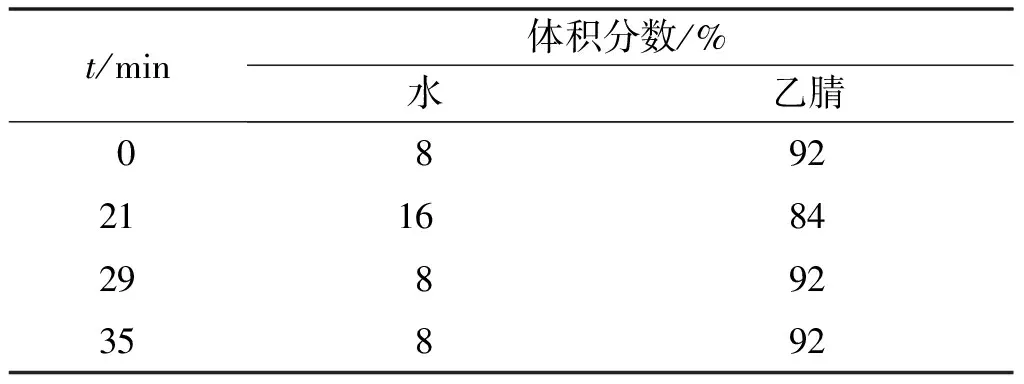
表1 线性梯度洗脱程序
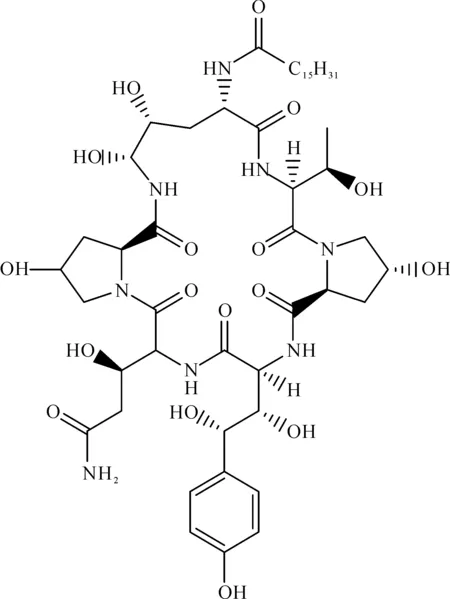
以纽莫康定C0对照品进行外标法定量,纽莫康定C0对照品溶液的配制与纽莫康定B0对照品溶液的配制相同。纽莫康定C0的结构式为
2.3 高压制备方法的确定
初始方法为鲁南新时代生物技术有限公司在50 mm高压制备液相(汉邦科技,制备型液相色谱系统,NS4000)上开发,是以二氯甲烷、甲醇和水体系制备纽莫康定B0,国内外未见具体报道。张宇等[11]采用了氯仿、甲醇和水体系,笔者以二氯甲烷进行实验,毒性低于氯仿[12],价格也低于氯仿,放大生产具有一定的优越性。初始方法:将纽莫康定B0粗品(质量分数为81.2%,HPLC检测如图1所示,纽莫康定B0的色谱纯度为88.58%、纽莫康定C0的色谱纯度为8.78%)用甲醇、二氯甲烷混合溶液(V(甲醇)∶V(二氯甲烷)=1∶4)溶解,波长276 nm;柱温20~30 ℃;流量70 mL/min;上样量1.7 g;流动相,V(二氯甲烷(工业))∶V(甲醇)∶V(水)=43∶9∶1;操作压力5~5.5 MPa。收集B0的HPLC色谱纯度>95%的制备液。纽莫康定B0的色谱纯度值和纽莫康定C0的色谱纯度值是经过面积归一化法得到的。以线性原则[13]放大到中试150 mm制备柱(Cs-prep150工业制备色谱系统,汉邦科技),流量调整为630 mL/min;进样量15.3 g;其他不变。在此基础上,对该方法进行了进一步优化。
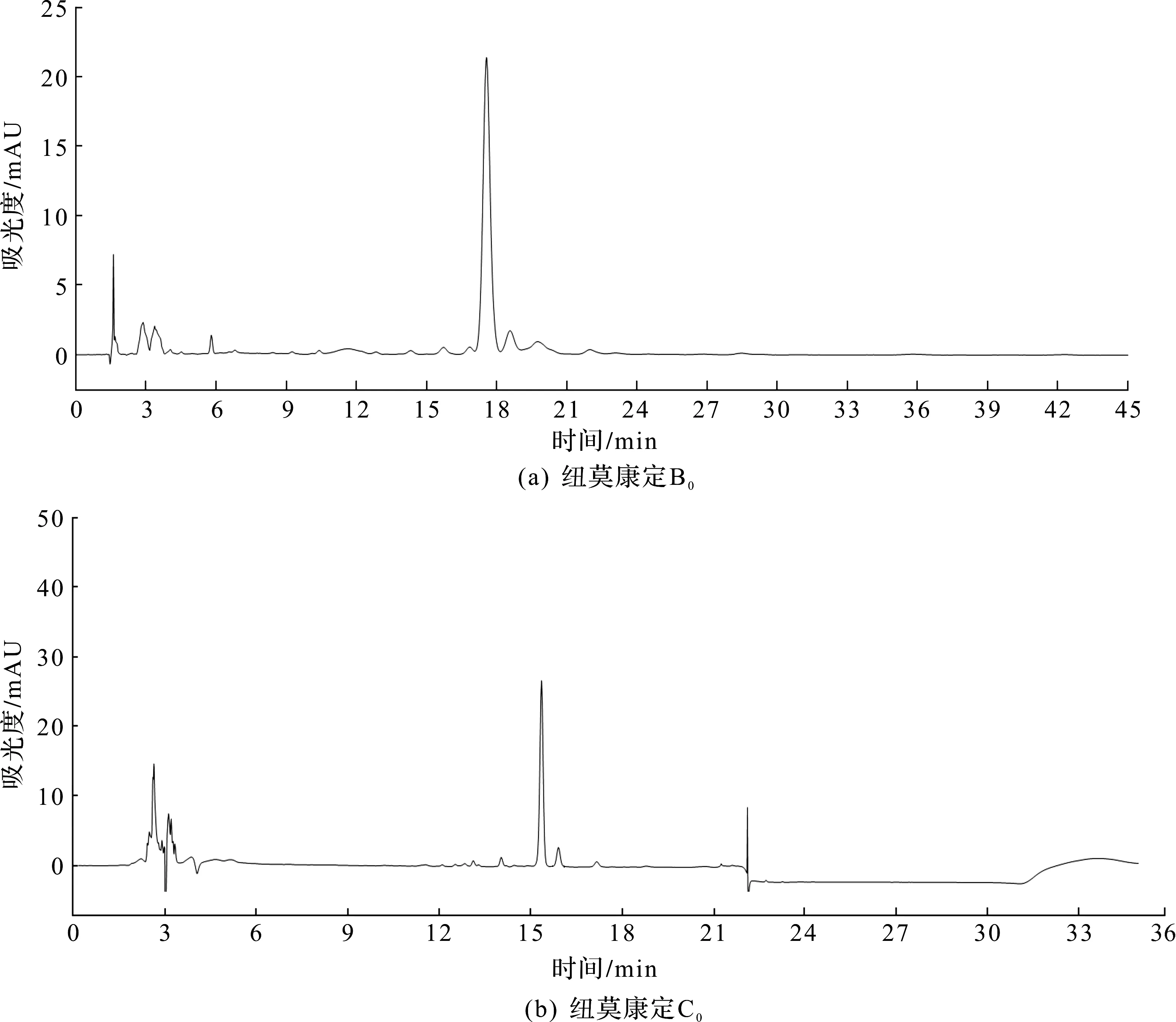
图1 纽莫康定粗品HPLC图谱
2.3.1 流动相比例的优化
根据正相色谱特点,通过调控二氯甲烷,对流动相比例进行优化。在初始方法中,实验分析了V(二氯甲烷(工业))∶V(甲醇)∶V(水)的5个比例对纽莫康定B0制备分离的影响,实验结果如表2所示。
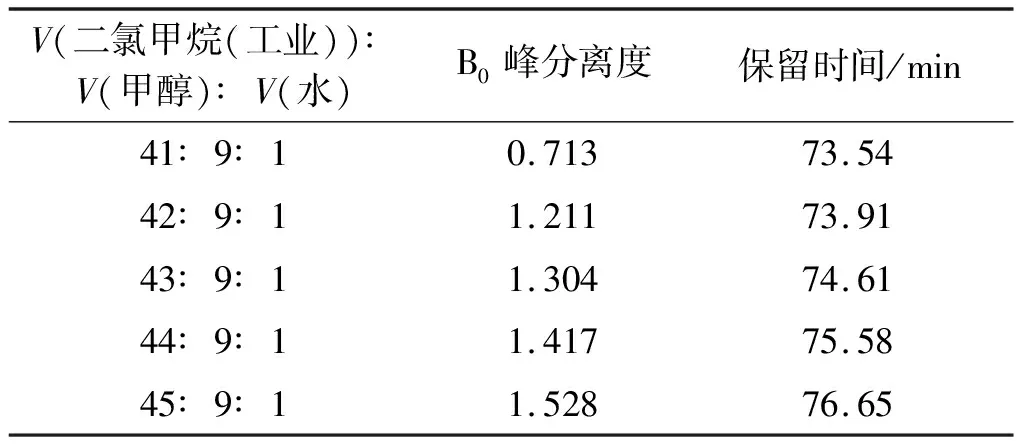
表2 流动相比例的筛选
由表2可知:随着二氯甲烷比例的增加,纽莫康定B0峰的保留时间逐渐增加,B0峰分离度逐渐增大,B0峰和相邻峰的重合越小。其中,41∶9∶1和42∶9∶1的流动相比例相比,后者B0分离度高于前者,且B0峰型较好,保留时间相差不大。保留时间越长,生产成本越大。综合考虑B0的分离效果和生产成本,故选择V(二氯甲烷(工业))∶V(甲醇)∶V(水)=42∶9∶1。
2.3.2 上样量的优化
上样量在色谱制备中影响着目的峰的分离度及制备效率。因此,在流动相比例确定为V(二氯甲烷(工业))∶V(甲醇)∶V(水)=42∶9∶1的基础上,实验选取了3种进样量(7.5,11.4,15.3 g)对纽莫康定B0制备的效果进行研究,实验结果如表3所示。
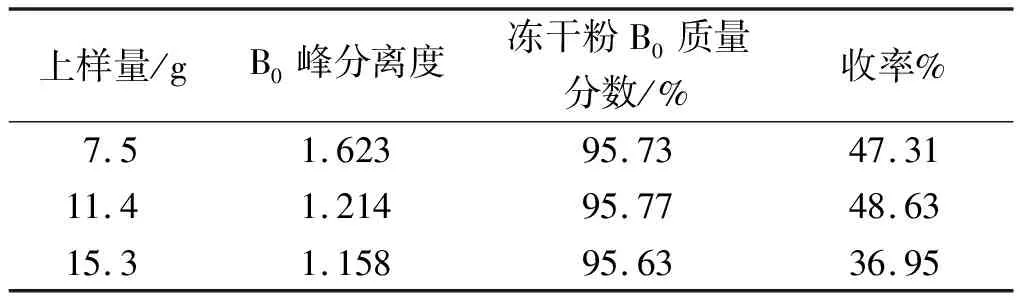
表3 上样量的筛选
上样量越大,分离度越小。对不同上样量下的B0峰进行分段收集,并对B0的HPLC色谱纯度大于95%的料液进行合并、减压浓缩和冻干,得到冻干粉的质量分别为3.01,4.7,4.8 g。3种冻干粉B0质量分数分别为95.73%,95.77%,95.63%,相差不大。计算收率分别为47.31%,48.63%,36.95%,其表达式为
式中:X为上样液对应粗品质量;Y为粗品B0质量分数。
由表3可知:上样量11.4 g收率高于上样量7.5,15.3 g。收率高,则在中试、大生产中预示着低成本、高效益,以及制药企业在市场中较高的竞争力。因此,综合考虑对纽莫康定B0的分离效果和收率,选择11.4 g上样量。
2.3.3 洗脱流速的优化
在高压制备液相色谱中,洗脱流速影响柱压、峰型,进而影响到制备效率及效果。故在确定流动相比例V(二氯甲烷(工业))∶V(甲醇)∶V(水)=42∶9∶1及上样量11.4g的基础上,考察了5个洗脱流速(480,530,580,630,680 mL/min)对纽莫康定B0制备的影响,结果如表4所示。
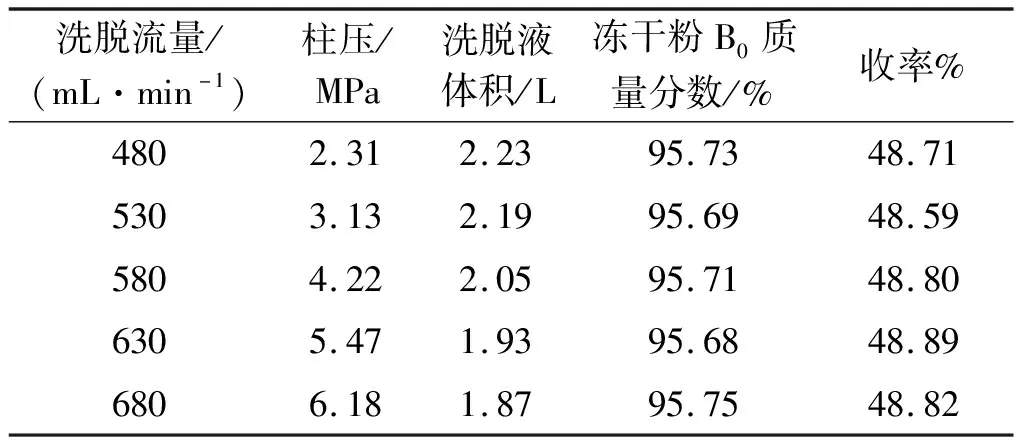
表4 洗脱流速的筛选
由表4知:洗脱流速越大,柱压越高,峰型越窄。对不同洗脱流速下的B0峰进行分段收集,并对B0的HPLC色谱纯度大于95%的料液进行合并、减压浓缩和冻干。流速越大,得到的洗脱合并液体积逐渐减小,且减小幅度不大。最终得到的冻干粉质量分别为4.71,4.70,4.72,4.73,4.72 g,其各数值相差很小。
随着流速的增加,柱压逐渐增加,而实验所用150 mm高压制备系统工作压力为0~7 MPa。随着进样次数的增加,柱压会逐渐增高,再生柱子会越频繁,如此会占用一定的工时和消耗再生溶剂,增加生产成本。另外,在5个流速下,冻干粉的收率和B0质量分数相差不大。综上考虑,优选480 mL/min的流速作为150 mm的制备柱流速。
2.4 纽莫康定B0的高压色谱制备
以优化后的高压制备条件,在150 mm制备柱上进行纽莫康定B0的制备。对B0峰分段收集,连续进样,合并纽莫康定B0的HPLC色谱纯度大于95%的制备液。将合并液进行减压浓缩,冻干,得到纽莫康定B0冻干粉。
3 结果分析
3.1 制备所得纽莫康定B0冻干粉质量分析
在优化后的150 mm制备条件(采用正相硅胶,V(二氯甲烷(工业))∶V(甲醇)∶V(水)=42∶9∶1,上样量为11.4 g粗品,洗脱流量为480 mL/min,检测波长分别为276,210 nm)下,上样量共980 g,得到4批次纽莫康定B0冻干粉质量共343.7 g,其质量情况如表5所示。
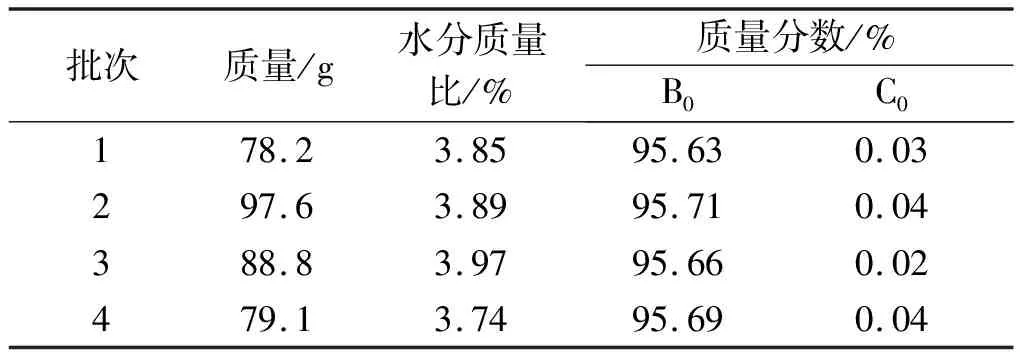
表5 4批次制备冻干粉质量
由表5知:各批次冻干粉B0质量分数均大于95%(以冻干粉1为例,其HPLC检测如图2所示,纽莫康定B0的色谱纯度为98.58%,纽莫康定C0的色谱纯度为0.04%),可用于下游合成。另外,各冻干粉C0杂质均降低至0.05%以下。周永正等[15]以表面修饰了极性基团的硅胶亲水材料进行层析,可以将关键杂质C0降至小于0.1%。相比,笔者开发的方法更能够对纽莫康定B0的异构体C0进行有效去除,纽莫康定B0是合成醋酸卡泊芬净的起始原料[16],因此在产品质量上,为下游合成提供了有效保证。
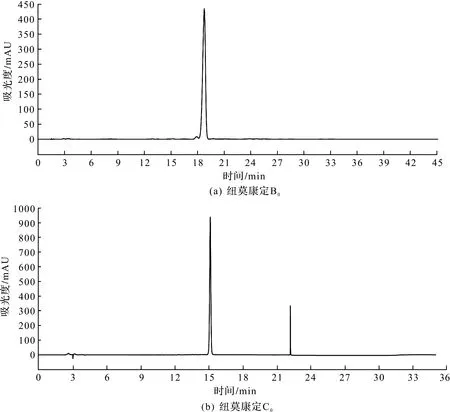
图2 纽莫康定制备冻干粉HPLC图谱
3.2 制备液稳定性分析
王欣荣等[17]发现分离过程中纽莫康定稳定性差的问题。在前期小试研究中,亦发现纽莫康定B0制备液常温放置不稳定,会有RRT1.36杂质峰出现。实验以纽莫康定B0制备收集液为研究对象,在4 ℃冷库(避光)的保存条件下对稳定性进行了简单摸索。液相检测数据如表6所示。由表6可知:4 ℃避光条件下,B0制备液放置7 d时,B0的HPLC色谱纯度降低了0.69%,RRT 0.96峰和RRT 1.14峰的HPLC色谱纯度基本不变;RRT 1.36峰在放置9 d时检测出来;13 d时,RRT 0.96峰的HPLC色谱纯度降低很小,纽莫康定B0的HPLC色谱纯度减小了0.71%,RRT1.14峰的HPLC色谱纯度增加了0.34%,RRT1.36峰的HPLC色谱纯度增加至0.27%。因此,纽莫康定B0制备液在4 ℃避光的条件下7 d内相对稳定。在中试生产中,纽莫康定B0制备液需及时放入4 ℃冷库中保存,需要7 d内处理完毕。在实际中试研究中,有4 ℃避光冷库作为辅助可以保证储藏条件,制备液放置时间为影响其稳定性的主要因素,因此实验中没有考察光照、温度对制备液稳定性的影响。
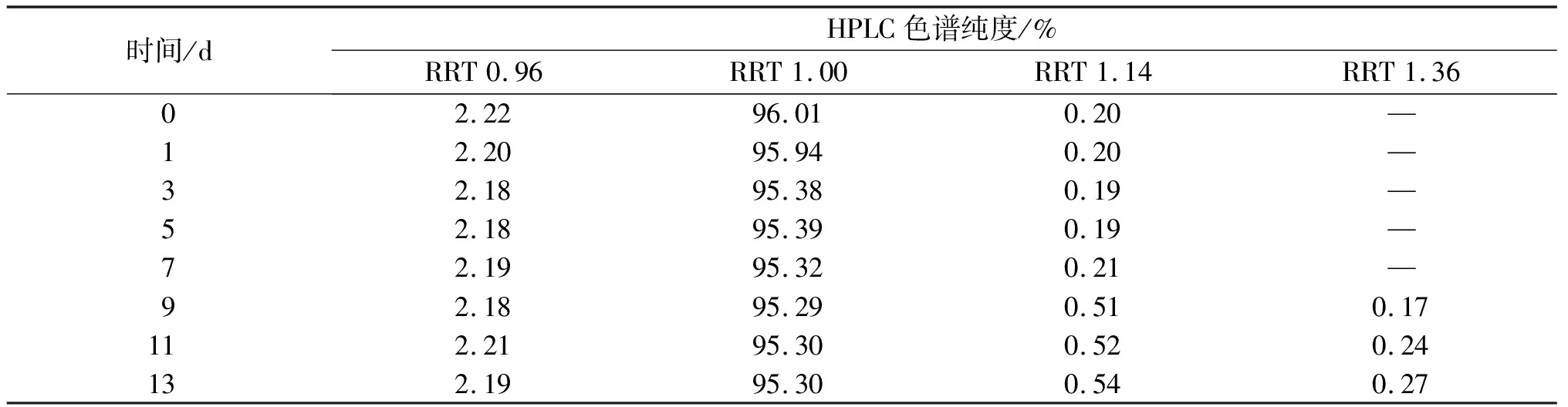
表6 纽莫康定B0制备液的稳定性
3.3 总收率分析
高压制备纽莫康定B0的中试研究分4个亚批进行,总消耗纽莫康定B0的粗品质量980 g。为了对放大规模的商业化生产提供数据支持和借鉴,需要对总收率进行计算,实验总收率计算式为
式中:E为冻干粉i的质量;F为冻干粉的B0质量分数。
经过计算,总收率为41.32%。影响总收率的因素主要有:填料、制备方法和卡泊芬净合成对纽莫康定B0的粗品质量需求等。为了提高总收率,可以和生产填料的厂商寻求合作,开发新型改良正相硅胶,进而继续优化适合其使用的色谱制备方法,优化卡泊芬净合成后的提纯工艺。亦或,统筹从发酵液至卡泊芬净整体的提取工艺,达到一个合理的平衡点,以降低其对纽莫康定B0的粗品质量要求,进而可以提高纽莫康定B0高压制备的收率。
另外,为了提高生产的总收率和降低成本,也可以将高压制备过程中产生的HPLC色谱纯度低于95%的B0制备液进行回收利用。具体方法:将低浓度制备液经过收集、浓缩和干燥后,得到回收冻干粉(HPLC检测如图3所示,纽莫康定B0的色谱纯度为84.30%,纽莫康定C0的色谱纯度为0.65%),再次进行制备回收套用。
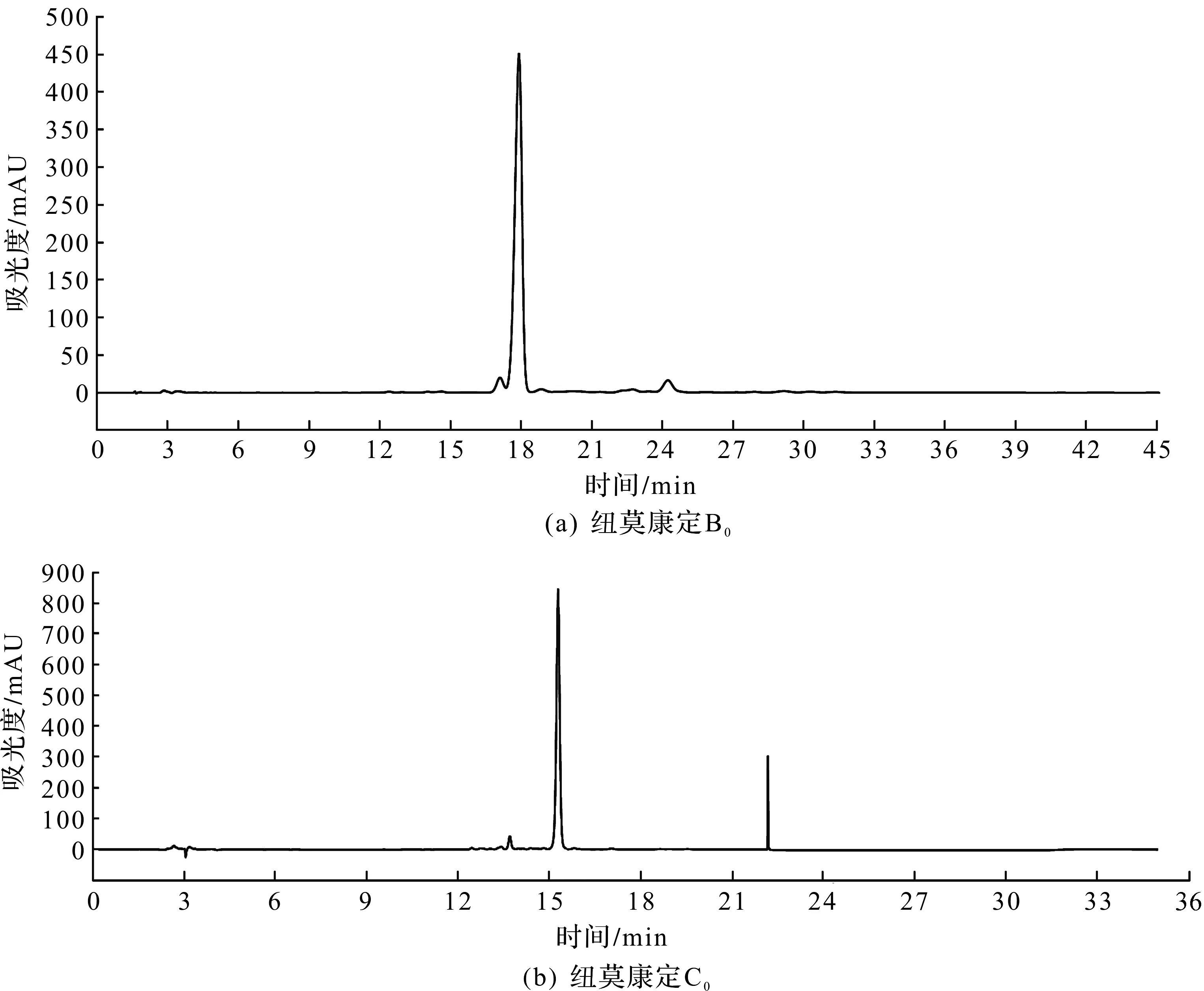
图3 回收冻干粉HPLC图谱
4 结 论
以二氯甲烷、甲醇和水体系制备纽莫康定B0,国内外未见具体报道。笔者以单因素实验的方法确定了150 mm高压制备纽莫康定B0的条件:采用正相硅胶,V(二氯甲烷(工业))∶V(甲醇)∶V(水)=42∶9∶1,上样量为11.4 g粗品,洗脱流量为480 mL/min,检测波长分别为276,210 nm。相比氯仿、甲醇和水体系,该制备工艺毒性小,更绿色环保。以优化后条件,可以得到质量分数95%以上的B0干粉,能有效将B0异构体C0降至0.05%以下,为下游合成卡泊芬净提供了更好的保证。通过对纽莫康定B0制备液稳定性研究,制备液需及时放入4 ℃冷库中保存,需要7 d内处理完毕,为进一步放大生产提供了数据支持。制备工艺生产周期短、成本低,且纯度高。
猜你喜欢
车迷(2022年1期)2022-03-29
云南化工(2022年2期)2022-03-18
现代艺术(2021年4期)2021-05-17
地震研究(2021年1期)2021-04-13
农药科学与管理(2021年2期)2021-03-16
科技与创新(2015年20期)2015-10-29
科技与企业(2015年20期)2015-10-21
